
by Jonathan Latham, PhD
On February 1st, 2020, Anthony Fauci, head of the US National Institute of Allergy and Infectious Disease (NIAID), secretly convened a group of select international virologists. Their task was to decide whether SARS-CoV-2, the virus newly emerged from Wuhan, was engineered.
Some key emails from their resulting discussions have only recently become available. In one, Robert Garry, a virologist from Tulane University, wrote to his associates: “I just can’t figure out how this gets accomplished in nature . . . it’s stunning.” Other emails show that many in the group agreed that the virus likely did not emerge naturally and most probably was altered in a lab.
Many months later, in August of 2020, Francis Collins, head of the US National Institutes of Health, emailed his predecessor, Nobel virologist Howard Varmus. Copying in Anthony Fauci, Collins wanted their opinions on a specific lab escape hypothesis, our Mojiang Miner Passage theory of the origin of the virus.
Taken together with the DEFUSE project, a US/China collaboration that proposed using US defense funding to take coronaviruses collected in China and give them ‘humanised” protein cleavage sites that would enhance their pathogenicity, such revelations are highly informative. They tell us that, even early on, many virologists, including the Fauci in-group, privately considered that a lab leak of an engineered wild coronavirus was plausible and some thought it likely.
It is striking then to find that, immediately after their discussions, many of the Fauci group led vociferous efforts to douse and denigrate any lab leak speculation, both in the media and in a string of subsequent scientific publications (Andersen et al., 2020; Holmes et al., 2021; Worobey et al., 2022; Pekar et al., 2022; Holmes, 2022) .
This article dissects some of these efforts to create an intellectually poisoned environment around the origin of the virus. Specifically, it is about how, amid the complications and confusions of emerging scientific information and journalistic revelations, this group of Fauci insiders has cynically and strategically attempted to dismiss a specific theory of COVID-19’s origin, the Mojiang Miners Passage Theory. This negative attention, as I will show by critiquing the views they express, embodies a paradox. It has arisen not because they see the theory as flawed but precisely because it is a highly viable and parsimonious explanation of SARS-CoV-2’s emergence.
Scientific Reputation management: the strategy
Amid the challenge of clear official disapproval of any lab leak discussion, only rarely has any virologist been willing to discuss publicly the detailed scientific merits of the different lab leak theories.
Instead, although university scientists are nowadays paid to ‘communicate science’, virologists with expertise relevant to the COVID-19 origin question have become notorious on the social media platform Twitter for the blocking of anyone, including scientists, engaging them in lab leak discussions.
In response, mocking Twitter accounts like BlockedByAVirologist have documented this suppression of scientific discussion. Our own account (@BioSRP), for example, has been blocked by prominent virologists Peter Daszak, Eddie Holmes, Kristian Andersen and Angela Rasmussen. Since our ‘offence’ each time was merely to draw polite attention to the science supporting a lab leak, it seems that this blockade is coordinated and tactically directed at anyone publicly discussing scientific alternatives to a natural zoonotic origin.
This should be strategically unsurprising. It is orthodox public relations that the advocates for a clear frontrunner (i.e. a natural zoonosis) should not publicly recognise a lesser-known rival, in this case a lab leak. The logic is that recognition only validates that rival and offers them free publicity.
With a few notable exceptions, the virological community has adopted this principle even in their scientific writings. Just a few days after they first convened, five members of the Fauci group submitted for publication an article titled “The proximal origin of SARS-CoV-2” (Andersen et al., 2020). This article was rapidly published. It categorically dismissed a Wuhan lab leak as a credible possibility.
Even though it is not even a full scientific paper (it is a “correspondence” piece) and its major conclusions were premature and some erroneous, this assessment nevertheless has 4453 citations (and still rising). By contrast, published hypotheses of COVID’s origins incorporating a lab leak all have very few citations. For example, the highly credible review paper “Might SARS-CoV-2 Have Arisen via Serial Passage through an Animal Host or Cell Culture?” has just 21 citations (Sirotkin and Sirotkin, 2020).
A more recent example of this scientific shunning is the paper: “The origins of SARS-CoV-2: A critical review”, published in September 2021 (Holmes et al., 2021). Despite claiming to present a “critical review”, the authors, many drawn once again from the Fauci group, do not authentically describe any lab leak hypothesis; nor do they cite even a single scientific paper or theory hypothesizing one (e.g. Segreto and Deigin 2020; Sirotkin and Sirotkin, 2020; Rahalkar and Bahulikar, 2020; Speciale, 2020; Kaina, 2021). Its readers are apparently expected to guess what a lab leak might have entailed.
Failing to cite competing theories or prior work is a basic transgression of scholarly ethics. On one level, citation failures are elementary, but the poet who wrote: ‘Oh what a tangled web we weave/When first we practice to deceive‘ was not wrong. Citation failure at scale implicates a broad web of peer reviewers, editors and journals. It is a key signal that virologists, as a profession, would rather defend risky virus research and the millions of dollars in biodefense and biosecurity money that it brings them, than stand up for science’s professed values of rigor, openness, and accountability.
For these reasons, a departure from this shunning strategy by a leader of the Fauci group ought to be treated as a significant event.
Andrew Rambaut attacks the Mojiang Miner Passage theory
In mid-October 2021, on the social media platform Twitter, virologist Andrew Rambaut, a Professor of Evolutionary Ecology at Edinburgh University in Scotland made an unusual intervention. He explicitly critiqued a lab leak theory of the origin of SARS-CoV-2 called the Mojiang Miners Passage theory (the MMP theory).

The MMP theory, which was conceived by biologist Allison Wilson and myself, is the proposition that the SARS-CoV-2 virus derives from one of six Chinese miners infected during a disease outbreak in 2012. The six miners, who had been shoveling bat guano, developed symptoms that closely resembled those of COVID-19 (Rahalkar and Bahulikar, 2020). Three of the six miners died and two suffered from chronic infections lasting over five months.
Officially, the outbreak remains unexplained. However, medical samples obtained from these miners were sent to the Wuhan Institute of Virology (WIV) where they were tested by the coronavirus research group of Dr Zheng-li Shi. According to three sources, these tests indicated the miners had infections from a novel coronavirus. Subsequent sampling in the same mine uncovered numerous bat coronaviruses, including RaTG13, which is very closely related to SARS-CoV-2 (Ge et al., 2016; Zhou P. et al., 2020).
Based on these observations, we have proposed that researchers at the WIV isolated a coronavirus from samples taken from at least one of the sick miners and that this coronavirus was studied and perhaps genetically manipulated there. We further proposed that, due to its long incubation inside a hospitalised miner, this virus had evolved to become highly human-adapted. It was this human-adapted virus, we proposed, that spilled over in Wuhan.
For Rambaut to critique the MMP theory directly is thus significant on several levels.
First, virologists’ stance of publicly ignoring specifics about lab leaks means that we have only an imperfect and superficial idea, embellished by occasional but out-of-date emails revealed through FOIA, (and more recently via congressional demands) what leading (and indeed most) virologists truly think about the origin of SARS-CoV-2 and why.
This lack especially applies to the Mojiang Miner Passage theory (MMP theory). Unlike lab leak theories that explain SARS-CoV-2 as largely or entirely the product of genetic engineering or other lab techniques, the MMP theory is largely biological in nature. Its’ evaluation relies heavily on estimating the plausibility of natural processes such as the infection, recombination, and evolution of viruses inside the body of a hospitalised miner. In contrast, determining the likelihood of lab engineering makes much greater use of written evidence from grants, emails, talks, and publications. The effect of this difference is that it is considerably more difficult for non-experts to independently evaluate the plausibility of the MMP theory than it is for them to do the same for other lab leak scenarios. Hence, assessing expert views is particularly critical.
For this reason, we welcomed the chance to debate (if only on Twitter) the merits of the MMP with a well-informed critic–and Andrew Rambaut is an especially prominent and well-connected one. Just since the pandemic began, he has accumulated over 40,000 citations (according to Google Scholar) for his coronavirus research, making him one of the world’s most cited scientists. Rambaut is additionally a founder of a virus research consortium called the ARTIC Network and he runs the popular website virological. He is also a founding member of the UK government’s SAGE advisory group on the pandemic.
More particularly, Rambaut is a leading proponent of a natural origin for SARS-CoV-2. He established this status very early in the pandemic as a co-author of the hugely influential “The proximal origin of SARS-CoV“, referred to above. This assessment was pivotal in suppressing initial concerns about a lab leak (but see also his co-authorship of Holmes et al., 2021 and Pekar et al., 2022). And perhaps most significant of all, Rambaut was one of the small group summoned by Dr Fauci.
Thus, Rambaut is one of the most significant scientists of the pandemic. Arguably, he is preeminent among its evolutionary virologists. For multiple reasons therefore, his views on COVID-19’s origin and the MMP theory are of exceptional interest.
A Twitter debate begins
Here is the point Andrew Rambaut chose to argue, on October 18th 2021:

This Oct 18th tweet is direct and emphatic. It conveys the impression that its author has a knock-down argument. As also can be seen from the screenshot, it repeats and amplifies an earlier tweet of his from Oct 11th. That Oct 11th tweet cited the @BioSRP account, which I administer.
The first point to notice about these tweets is the apparent trigger, which may explain why Rambaut broke ranks to attack our theory.
In his two tweets Rambaut alluded to the “elevated rate of evolution” seen in “chronic infections”. This refers to a fascinating but also alarming recent scientific discovery–about which I had just spoken to a workshop on the origins of SARS-CoV-2 hosted by the British Medical journal. This discovery, which may well hold the key to the future of the pandemic, has been much discussed by experts but mostly out of public sight.
The basic observation is that, with the exception of Delta, almost every novel variant worthy of being named by the WHO, including Alpha (from the UK), Beta (from South Africa), Gamma (from Brazil), Mu (from Colombia), and mostly recently Omicron, has emerged according to a pattern that is consistent but entirely unexpected (see e.g. Faria et al., 2021).
Viral variants are so called because they contain large numbers of mutations, many of which are unique to each of them. This makes them radical outliers from the wider population of viruses. Second, they are extremely successful, i.e. they are remarkably well-adapted to their human hosts. Third, and most surprising, their nearest known relatives are not, as one might suppose, other contemporaneous SARS-CoV-2 strains, but instead viruses that became extinct many months (or even years) before. The Alpha variant, for example, emerged in September, 2020, from a virus previously seen in about April, 2020; Omicron emerged in November 2021 from a virus last seen in about May 2020 (Hill et al., 2022). Thus, each variant is hyper-adapted but simultaneously a genetic throwback (Stern et al., 2021; Martin et al., 2022).
This can be seen in Figure 2. It is adapted from Laiton-Donato et al. who characterised in detail the variant known as Mu (B.1.621). Their figure also includes variants Alpha (B.1.1.7), Beta (B.1.427), and Gamma (P.1.). The unusual evolution of all these variants is signalled by the exceptionally long unbranched line leading to each variant and emanating from the centre of the chart (which is the virus origin). In this figure, since a spot represents a sampling event, long lines between spots show rapid evolution and lack of branching indicates genetic isolation.
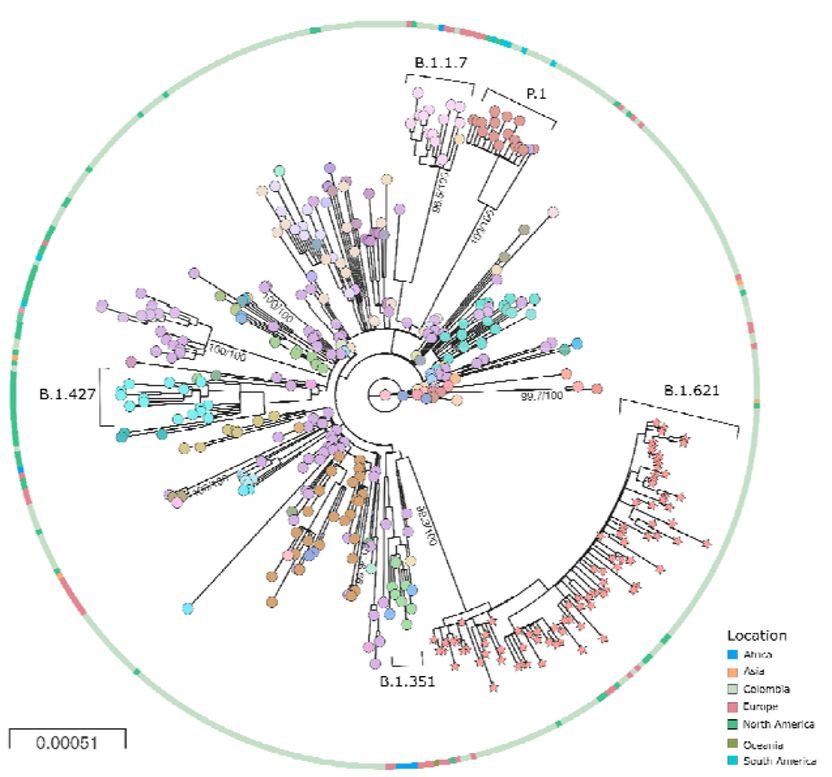
Since Alpha, Beta, Omicron and other novel variants have successively outcompeted each other as well as other virus strains, they have, in effect, dictated the course of the pandemic. Given this pattern, others can be expected to emerge in the near future. Thus, a quiet but high stakes debate is happening, asking what is the origin of these genetic freaks?
One proposal is that novel variants originate in human populations that are epidemiologically isolated (or at least hidden from sequencing projects). Others have suggested that the virus spread into another species (such as a mouse) where they evolved over a long period before jumping back into humans (Martin et al., 2022; Hill et al., 2022).
But the explanation favoured by most virologists, including Rambaut, is that these variants have emerged from chronically infected individuals who were either hospitalised or immunocompromised (Hill et al., 2022). It is theorised that these individuals acquired SARS-CoV-2 infections early in the pandemic and, in their vulnerable state, failed to eradicate the virus. Long incubation inside a single infected patient allowed evolution to occur at an unusually rapid pace, leading ultimately to the creation of radically different versions of SARS-CoV-2. Eventually, these patients transmitted their novel virus, thereby seeding the global spread of a new variant.
This idea is strongly supported by multiple publications describing patients with chronic SARS-CoV-2 infections who accumulate large numbers of adaptive mutations (Avanzato et al., 2020; Kemp et al., 2021; Choi et al., 2020; Corey et al., 2021; Truong et al., 2021).
This chronic infection theory accounts for how novel variants can be so different from currently circulating strains and simultaneously so successful, especially at evading immunity, and yet go undetected for months or years despite mass sampling and genome sequencing.
For us, the accumulating evidence for accelerated evolution in chronically infected patients has extra significance. When we first proposed, as part of the MMP theory, that chronic coronavirus infections of the miners facilitated a large evolutionary leap in which a bat virus (or viruses) evolved into a human one, some virologists claimed that such rapid evolution, perhaps spanning many hundreds of nucleotides, was implausible inside a single individual.
The appearance of Alpha, Omicron, and other variants means that the emergence of SARS-CoV-2 in a single chronically infected miner can no longer be dismissed for want of evolutionary plausibility.
If our MMP theory is correct, the same biological phenomenon that creates most variants, also created the virus itself.
Hence also Rambaut’s concern, as a leading author of papers identifying and analysing these new variants, about the MMP theory (e.g. Hill et al., 2022).
Why analyse a Twitter debate?
At first sight, dissecting arguments made on social media, even those from a renowned scientist, might seem of doubtful value. However, examining Rambaut’s thinking, which he went on to elaborate as we debated, proved highly worthwhile.
First, his tweets are sufficient to expose profound weaknesses in his arguments. Rambaut repeatedly misrepresented our views and so ended up critiquing us for positions we don’t hold. Rambaut also critiqued us on points that are ultimately of little consequence. Even if we were wrong, it therefore would be of negligible consequence. Finally, Rambaut’s expression “because phylogenetics” (see the tweet above) is baby talk. It is an unsubtle signal, reiterated in some of his subsequent tweets, to dismiss us as scientific naifs. It’s a form of the classic ad hominem attack. When you don’t like the message, shoot the messenger.
Nor are these errors his alone. Similar ones have been made by others. Indeed, many apparently intentional, misconceptions have been spread in the media about the mine and the miners.
What is special to Rambaut’s errors, however, is this: Rambaut is clearly a highly competent and experienced scientist. Any competent person in possession of a powerful argument does not bother to debunk straw men, critique details, or resort to ad hominem attacks. His deployment of such dubious tactics strongly suggests that Rambaut has no genuine case against the MMP theory.
Could it be that the scientific community of which he is a leader has kept from him its best arguments against the MMP theory? This is not likely. More probable, given that no coherent refutation of the MMP theory has yet been presented by anyone, is that none of the lab leak critics possesses decisive arguments against it (Frutos et al., 2021). If true, this is a very powerful observation.
Rambaut misrepresents the MMP theory
The second point to notice about the tweets above is the considerable effort that went into making them, resulting in an extended thread of tweets, including figures.
The third is that these two initial tweets both mis-describe the MMP theory. Based on this mis-characterisation Rambaut then ‘debunks’ our thinking: how ridiculous, he suggests, for us to claim that SARS-CoV-2 could have arisen directly from RaTG13, the SARS-CoV-2-like bat virus found in the mine.
We were away from the internet when Rambaut first tweeted these and so had no opportunity to reply.
Meanwhile, this ‘debunking’ gathered over a hundred re-tweets, many from professional virologists. However, after the second tweet, we did respond.
Clarifying the role RaTG13 did play in the evolution of SARS-CoV-2
First identified in 2013, RaTG13 is a SARS-related bat coronavirus found in the Mojiang mine. At the outset of the pandemic, although it differs from SARS-CoV-2 by about 1140 nucleotides, it was the most closely related wild coronavirus to SARS-CoV-2 known (Ge et al., 2016; Zhou P. et al., 2020). This greater similarity compared to any other wild coronavirus held true along the whole length of its genome, which is almost 30,000 nucleotides long.
However, since the pandemic began, intensive searching for other wild coronaviruses has unearthed 19 other viruses (most are from bats but a few are from smuggled pangolins) that are also closely related to SARS-CoV-2 (Zhou P. et al., 2020; Lam et al., 2020; Chan and Zhan 2020; Temmam et al., 2022; Murakami et al., 2021; Delaune et al., 2021; Wacharapluesadee et al., 2021; Zhou H. et al., 2021; Li L. et al., 2021). Together with SARS-CoV-2, these viruses form a distinct evolutionary clade, sometimes called the nCoV clade (Lytras et al., 2021). Only one of these viruses, BANAL-52, is closer in overall nucleotide similarity to SARS-CoV-2 than RaTG13 (Temmam et al., 2022).
The most recently discovered of these close relatives are seven bat viruses (two are incomplete virus sequences) from northern Laos (Temmam et al., 2022). These Laotian viruses are named BANAL-27, BANAL-52, BANAL-103, BANAL-116, BANAL-236, BANAL-242 and BANAL-247.
Genome comparisons by these latter authors, of all the members of the nCoV clade, including these newest viruses, let them estimate which among these 19 wild coronaviruses have extensive segments of their genomes that are statistically closer in sequence to human SARS-CoV-2 than the others. Five of the 19 viruses can make a clear claim to be closest for at least one stretch of its genome: BANAL-52, BANAL-103, RaTG13, and two viruses from S. Yunnan called RmYN02 and RpYN06 (see Temmam et al., Figure 2). Regardless of whether one favours a lab leak or a natural origin, these five viruses collectively represent the most plausible currently known ancestors for the backbone of SARS-CoV-2.
A key finding of this work is that SARS-CoV-2 is therefore a mosaic genome. It was created through genetic recombination and no single currently known virus is its one true ancestor. A second implication is that all 19 members of the clade have a similarly complex history of recent and ancient recombination-they are all mosaics. In retrospect, this is not totally surprising given that coronaviruses recombine extremely readily (Makino et al., 1986).
A third crucial inference of this analysis is that the five most plausible candidates for its parent all come from bats and from a quite small geographic region spanning southern Yunnan, in China, and its neighbour, northern Laos. The longest axis of this region is 500 Km.
This geographic concentration of all its closest relatives is also of high consequence for understanding the origin of SARS-CoV-2. It means that S. Yunnan/N. Laos, is where a wild virus closely related to SARS-CoV-2 likely exited (by whatever means) its bat host. Assuming no human intervention, the jump from a bat established the virus in a new host species (either a human host or another animal) on its way to becoming SARS-CoV-2 itself. Very importantly, this region where the initial jump from bats occurred includes the Mojiang mine. Much of this is detailed in an article we wrote, in August 2021, on the phylogeography of newly discovered bat coronaviruses.
As our August 2021 article made very clear, as a consequence of these new data points, neither we nor anyone else currently considers that SARS-CoV-2 evolved from RaTG13 ‘directly’ (i.e. that RaTG13 is the only parent).
So, while Rambaut’s tweets, and even more so the thread below them, imply that we currently believe SARS-CoV-2 is directly descended from RaTG13, we do not. His supposed debunking is thus just an attack on a straw man.
For the record, a long time ago, in July 2020, when we first elaborated our Mojiang Miner Passage theory, we did propose that RaTG13 was the direct ancestor since, at the time, it was the closest known relative and there was then no clear evidence for recombination between viruses. But we were also careful to leave room in the text (in several passages) for the subsequent discovery of viruses more closely related to SARS-CoV-2. For example, we noted that any virus present in the mine could have contributed genetic material to SARS-CoV-2. We also noted that, when the mine was sampled after the outbreak, it contained numerous coronaviruses and any of these (or other unsampled ones) could have recombined to contribute to the evolution of SARS-CoV-2.
And Rambaut may have forgotten that, just two weeks after our MMP proposal appeared (in July 2020), he himself co-authored a paper reaching the identical conclusion–that, given the data then available, RaTG13 was the apparent direct ancestor:
” SARS-CoV-2 itself is not a recombinant of any sarbecoviruses detected to date.”
So, at that time, direct descent of SARS-CoV-2 from RaTG13 was the standard and most parsimonious view. Rambaut even held it himself.
But there is a more important logical flaw in Rambaut’s tweets. It is their implication that the question of direct or indirect descent is even important. The key elements of the MMP theory are that 1) the miners were infected with bat coronaviruses acquired in the mine and 2) that a humanised coronavirus evolved inside them and was brought to Wuhan in the medical samples taken from them. So, had we hypothesised incorrectly precisely which coronavirus isolate(s) infected the miners, it would not matter–such an error would not refute the theory itself. Rambaut’s ‘direct descent’ criticism is thus doubly faulty. It is wrong and irrelevant.
Nevertheless, Rambaut is a black belt in virus evolution. Phylogenetics is his full-time profession. He even invents the software with which it is done. Consequently, it seemed unlikely that he would be entirely ignorant of his errors. So, I felt it was important to push him. Maybe, if prodded, he would produce a valid criticism of the MMP? Or maybe he would make more revealing errors? And, if he truly had solid criticisms, we could improve or discard our theory as appropriate.
If, on the other hand, he had none, then Andrew Rambaut, the arguably best-informed and most respected denier of lab leak origin theories, would stand exposed. A leading scientist exposed as incapable of substantiating his glib online dismissals. Similarly, his repeated ignoring of plausible lab leak theories in high-profile papers like the recent ‘critical review’ of COVID-19’s origins would be unjustifiable (Holmes et al., 2021). He would be guilty of very bad faith indeed.
Andrew Rambaut introduces a second straw man
As a first step, we quickly tweeted back:
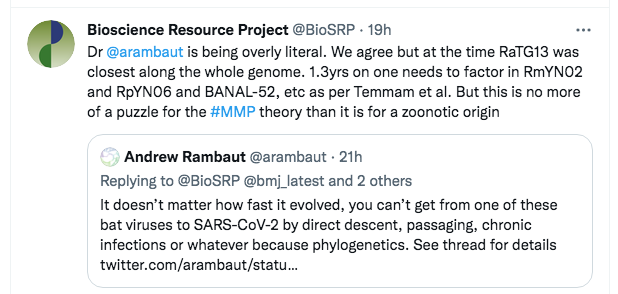
The tweet was primarily to indicate what we considered was his primary mistake, but the reference to Temmam et al was also a signal that we knew the evidence base, including the newest discoveries from Laos.
A few minutes later, Rambaut replied with a somewhat different argument:
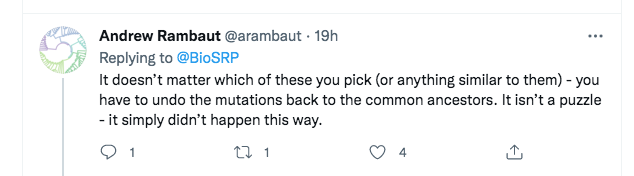
However, this too is a straw man. What Rambaut is saying here is that even if one considers the other viruses in the nCoV clade, none of them is a true ancestor of SARS-CoV-2. So Rambaut was at least accepting that we were not defending direct descent from RaTG13. Instead, he was transitioning to a different line of attack, that “SC2” (SARS-CoV-2) can’t have been created even from a “combination of viruses we know about”.
To understand the plausibility of Rambaut’s new point it helps to consider the subtleties of the specific wording Temmam et al used to describe their own proposed evolutionary history of SARS-CoV-2. Their phrasing is slightly awkward, but for a purpose.
They wrote (note below that R. malayanus, R. pusillus and R. affinis are the Rhinolophid bat species these viruses were found in):
“SARS-CoV-2 presents a mosaic genome, to which contributed more than five sequences close to sequences published or determined during this study: R. malayanus RmYN02 and R. pusillus RpYN06 viruses found in China in 2019, R. affinis RaTG13 coronavirus found in China in 2013, and R. malayanus BANAL-52 and R. pusillus BANAL- 103 found in North Laos in 2020 (this study).” (Temmam et al., 2022)
The passage states that SARS-CoV-2 is a mosaic but also that it is not simply a mosaic of the viruses we know of. The reason for making this distinction is that the exact viruses we know of probably didn’t exist at the time either of the spillover or when the various recombination events leading to SARS-CoV-2 occurred. This is why Temmam et al. carefully added the caveat “close to”–it prevents anyone mistaking their mosaic suggestion for a simplistic cut-and-paste type scenario.
So, as a teaching point, it is valid for Rambaut to point out that SARS-CoV-2 cannot be a simple cut-and-paste mosaic. But no one with even a moderate understanding of virus evolution is “imagining” this. Certainly not ourselves. Once again, Rambaut has taken aim at a straw man. Once again too, Rambaut takes the ad hominem option: imputing naiveté on our part.
For his next move, Rambaut changed tack again. He proposed that there is no need for the MMP theory:

Here, we disagree again. The first part of my reply below points out that, even aside from the origin of the backbone of the virus, there are multiple ongoing mysteries pertaining to the origin of SARS-CoV-2 that the MMP theory can solve:
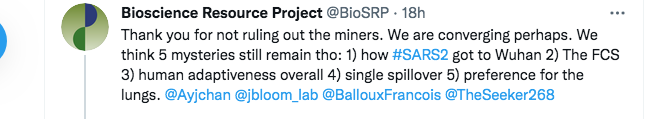
(FCS is shorthand for the Furin Cleavage Site. This is the short protein cleavage motif crucial to human infection and whose surprising presence in SARS-CoV-2 has caused the Fauci group, David Baltimore, and many others, to doubt a natural origin).
Nevertheless, the main focus of our exchange was the provenance of SARS-CoV-2’s backbone; and, by saying there was “no need” for the MMP theory, it seemed there was a genuine major difference between us.
When we initially formulated the MMP theory, the genetic gap between SARS-CoV-2 and its nearest known relative (at the time RaTG13) was 1140 nucleotides (3.8% of the SARS-CoV-2 genome). If, in line with Temmam et al., we assume that SARS-CoV-2 is a mosaic of viruses close to BANAL-52, BANAL-103, RaTG13, RmYN02, and RpYN06, then the genetic gap from SARS-CoV-2 to its wild relatives has shrunk to around 800 nucleotide differences (i.e. 2.4% of the SARS-CoV-2 genome).
Not all of this gap has to evolve in the miners. It is normal, when thinking about evolution at this level of detail, to assume the existence of an entity, in this case a virus, known as the Most Recent Common Ancestor (MRCA). For example, no evolutionist assumes that humans descended directly from chimps or gorillas (nor vice versa); rather, we evolved from a hypothetical predecessor species that perhaps resembled humans and chimpanzees equally. This predecessor is the MRCA of chimps and humans. An MRCA implies that, of the nucleotide differences between chimps and humans, some accumulated as the MRCA evolved into a chimp and the rest accumulated during the transition between the MRCA and humans. Other things being equal, half of the mutations by which any two species differ will lie on each branch leading from the MRCA.
If the MMP theory is true, the branch point likely occurred at or before infection of the miners. Hence, around 400 nucleotide mutations (half of 800) likely occurred in the miners. In principle, the same span could also have been bridged during passaging in lab animals and cell cultures or, as Rambaut prefers, as part of a natural zoonosis. Whichever, it is unclear why 400 nucleotides, which is over 1% of the virus genome, should not be considered a sizeable evolutionary gap to be filled.
Our Twitter exchange continued on a while longer. It can be seen in full here; (it could, until Andrew Rambaut deleted his account); but the essential point is that Andrew Rambaut never provided a clear or substantive refutation of the MMP theory.
For all his phylogenetic maneuvering and aggressive language, he fired only blanks and his targets were straw men and red herrings.
Given his expertise as the grand expert on coronavirus evolution, this absence of a substantive argument is hugely significant. Apparently, Andrew Rambaut, a founding member of the Fauci in-group and prolific author of high-profile analyses concluding that COVID-19 has a natural origin, knows no decisive scientific or biological argument against the MMP theory. Nevertheless, he was willing to loudly denounce it with faux arguments.
Other criticisms of the MMP theory
Still, the ultimate significance of all this for the MMP theory depends also on there not being other scientific challenges that refute it, ones that are perhaps outside of Rambaut’s expertise. There are two such potential challenges.
The first is the claim by the WIV that no coronavirus was ever detected in the miners’ serum samples, an the assertion first made in the addendum to Zhou P. et al., (2020). The second claim is that the miners had a fungal infection. Close inspection of both claims shows that they too are seriously flawed.
In the case of the proposed fungal infection, there is first the fact that the claim of a fungal infection has (at least in English) never been made directly by Zheng-li Shi of the WIV or anyone else in a position to know. Instead, the claim was initially made on her behalf by journalist Jane Qiu, who interviewed Zheng-li Shi for Scientific American. This indirectness would not matter if actual scientific evidence was offered in its support, but it never has been. The obvious place to provide this support was the Zhou addendum noted above, yet WIV scientists provided none.
As for what virus, if any, was detected in the samples taken from the miners, Dr Shi and her co-authors have only ever made one written claim, which is in that addendum. The statements in it are critical since they have been widely taken as a refutation of the MMP theory.
However, careful reading of the addendum reveals no direct denial that a coronavirus was found. Instead, the addendum merely states that the results of specific tests on specific samples yielded negative results. The central question–whether or not the miners were infected with any novel coronavirus, is thereby dodged. Moreover, contrary to all scientific norms, the methods used for the experimental tests that were performed on the miners’ samples are not described and their results are not shown.
It is both relevant and legitimate to raise this because WIV scientists have a history of omitting crucial scientific information. In their first ever publication on SARS-CoV-2 (Zhou P. et al., 2020), Zheng-li Shi’s group failed to mention that RaTG13 was obtained at the site of a disease outbreak (i.e. the Mojiang mine) and the authors also implied, falsely, that RaTG13 had only just been sequenced and studied. In the same paper, they also failed to note the presence of a furin cleavage site in SARS-CoV-2. This omission has been noted before, but it looks much stranger now, in the light of their own DARPA grant proposal (recently released) to add furin cleavage sites to wild coronaviruses. These three facts, had they been revealed at the time, would doubtless have transformed early discussions of COVID-19’s origin.
The indirectness of the addendum is a crucial matter. As we have previously reported, three other scientific sources, whose veracity is not in doubt (the Masters’ thesis of Li Xu, the PhD thesis of Canping Huang, and George Gao, head of China’s CDC), all assert that tests done at the WIV did find evidence of a coronavirus infection in samples from the miners.
Very recently, the same Jane Qiu has claimed on behalf of Dr Shi, that the discrepant conclusions arose from false positive results. Here again, identical caveats apply; Dr Shi is not quoted directly and no scientific evidence is presented for the claims. For example, how was it concluded that these results were false positives? Can the data or the samples not be shared? If there was no indication of coronavirus infection in the miners, why did so many teams of virologists, especially corona-researchers, sample the mine, especially its bats, so intensively and for many consecutive years thereafter?
These and many other questions remain, casting doubt on almost every aspect of the WIV’s account of the mine outbreak.
Ultimately, in the eighteen months since we first proposed it, literally no-one has managed to find a clear or substantive reason to dismiss the Mojiang Miners Passage theory. Even the strongest criticisms of it are, at best, unsubstantiated. The rest are ad hominems or irrelevant.
Instead, all of the evidence that has subsequently emerged is either consistent with the theory that SARS-CoV-2 evolved in the miners or has reinforced it.
For example, the MMP theory implies a bat-to-human spillover with no intermediate animal. For this reason it is noteworthy that still no mammalian intermediate host has been identified. What has been found are viruses from Northern Laos that are highly related to SARS-CoV-2 in the receptor binding domain of the spike protein and that also infect human cells with high efficiency. This shows the existence of naturally occurring spike proteins–not far from the mine–that are competent to jump directly into humans; and a jump directly into human cells is what the MMP theory relies on.
The MMP theory therefore remains fully viable. It is not only entirely plausible, it is the single most parsimonious explanation for the principle tasks of every COVID origin theory, which are to explain a single spillover, in Wuhan, of a highly human-adapted SARS-related coronavirus 1700 Km from its natural home.
As we recently demonstrated, this situation is not changed at all by two recent preprints that were widely but incorrectly construed in the media as confirming that the Huanan market was the site of several natural spillovers from an unknown host (Pekar et al., 2022; Worobey et al., 2022).
And who are the senior authors of these two papers (Pekar et al., 2022 and Worobey et al., 2022) who yet again attempt to shoehorn ill fitting data into a zoonotic theory? It should by now be no surprise that the Worobey senior authors are Holmes, Garry, Andersen and Rambaut–all members of the original Fauci in-group and Pekar et al. is authored by these four plus Michael Worobey and Joel Wertheim.
Over two years after SARS-CoV-2 first emerged in Wuhan, we can perceive that Fauci’s COVID origin in-group truly has developed into a S.W.A.T team, in this latter case battling against all lab origin theories (see also Harrison and Sachs, 2022).
References
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature medicine, 26(4), 450-452.
Avanzato, V. A., Matson, M. J., Seifert, S. N., Pryce, R., Williamson, B. N., Anzick, S. L., … & Munster, V. J. (2020). Case study: prolonged infectious SARS-CoV-2 shedding from an asymptomatic immunocompromised individual with cancer. Cell, 183(7), 1901-1912.
Boni, M. F., Lemey, P., Jiang, X., Lam, T. T. Y., Perry, B. W., Castoe, T. A., … & Robertson, D. L. (2020). Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature microbiology, 5(11), 1408-1417.
Chan, Y. A., & Zhan, S. H. (2020). Single source of pangolin CoVs with a near identical Spike RBD to SARS-CoV-2.
Choi, B., Choudhary, M. C., Regan, J., Sparks, J. A., Padera, R. F., Qiu, X., … & Li, J. Z. (2020). Persistence and evolution of SARS-CoV-2 in an immunocompromised host. New England Journal of Medicine, 383(23), 2291-2293.
Corey, L., Beyrer, C., Cohen, M. S., Michael, N. L., Bedford, T., & Rolland, M. (2021). SARS-CoV-2 variants in patients with immunosuppression. New England Journal of Medicine, 385(6), 562-566.
Delaune, D. et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat. Commun. 2021 121 12, 1–7 (2021) doi: 10.1038/s41467-021-26809-4.
Faria, N. R., Mellan, T. A., Whittaker, C., Claro, I. M., Candido, D. D. S., Mishra, S., … & Sabino, E. C. (2021). Genomics and epidemiology of the P. 1 SARS-CoV-2 lineage in Manaus, Brazil. Science, 372(6544), 815-821.
Frutos, R., Javelle, E., Barberot, C., Gavotte, L., Tissot-Dupont, H., & Devaux, C. A. (2022). Origin of COVID-19: Dismissing the Mojiang mine theory and the laboratory accident narrative. Environmental Research, 204, 112141.
Ge, X. Y., Wang, N., Zhang, W., Hu, B., Li, B., Zhang, Y. Z., … & Wang, B. (2016). Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virologica Sinica, 31(1), 31-40.
Harrison Neil L. and Sachs Jeffrey D. (2022) A call for an independent inquiry into the origin of the SARS-CoV-2 virus. PNAS 119 ( 21 ) e2202769119 https://doi.org/10.1073/pnas.2202769119
Hill, V., Du Plessis, L., Peacock, T. P., Aggarwal, D., Colquhoun, R., Carabelli, A. M., … & Rambaut, A. (2022). The origins and molecular evolution of SARS-CoV-2 lineage B. 1.1. 7 in the UK. bioRxiv.
Holmes, E. C., Goldstein, S. A., Rasmussen, A. L., Robertson, D. L., Crits-Christoph, A., Wertheim, J. O., … & Rambaut, A. (2021). The origins of SARS-CoV-2: a critical review. Cell.
Hu, B., Zeng, L. P., Yang, X. L., Ge, X. Y., Zhang, W., Li, B., … & Shi, Z. L. (2017). Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS pathogens, 13(11), e1006698.
Kaina, B. (2021) On the Origin of SARS-CoV-2: Did Cell Culture Experiments Lead to Increased Virulence of the Progenitor Virus for Humans? In Vivo 35 (3) 1313-1326; DOI: https://doi.org/10.21873/invivo.12384.
Kemp, S. A., Collier, D. A., Datir, R. P., Ferreira, I. A., Gayed, S., Jahun, A., … & Gupta, R. K. (2021). SARS-CoV-2 evolution during treatment of chronic infection. Nature, 592(7853), 277-282.
Laiton-Donato, K., Franco-Muñoz, C., Álvarez-Díaz, D. A., Ruiz-Moreno, H. A., Usme-Ciro, J. A., Prada, D. A., … & Mercado-Reyes, M. (2021). Characterization of the emerging B. 1.621 variant of interest of SARS-CoV-2. Infection, Genetics and Evolution, 95, 105038.
Lam, T. T. Y., Jia, N., Zhang, Y. W., Shum, M. H. H., Jiang, J. F., Zhu, H. C., … & Cao, W. C. (2020). Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature, 583(7815), 282-285.
Li X., 2013 The Analysis of 6 Patients with Severe Pneumonia Caused by Unknown viruses. (MSc)
Li, L. L., Wang, J. L., Ma, X. H., Sun, X. M., Li, J. S., Yang, X. F., … & Duan, Z. J. (2021). A novel SARS-CoV-2 related coronavirus with complex recombination isolated from bats in Yunnan province, China. Emerging microbes & infections, 10(1), 1683-1690.
Lytras, S., Hughes, J., Martin, D., de Klerk, A., Lourens, R., Pond, S. L. K., … & Robertson, D. L. (2021). Exploring the natural origins of SARS-CoV-2 in the light of recombination. bioRxiv.
Makino, S. H. I. N. J. I., Keck, J. G., Stohlman, S. A., & Lai, M. M. (1986). High-frequency RNA recombination of murine coronaviruses. Journal of Virology, 57(3), 729-737.
Martin, D. P., Lytras, S., Lucaci, A. G., Maier, W., Gruning, B., Shank, S. D., … & Pond, S. L. K. (2022). Selection analysis identifies unusual clustered mutational changes in Omicron lineage BA. 1 that likely impact Spike function. bioRxiv.
Murakami, S., Kitamura, T., Suzuki, J., Sato, R., Aoi, T., Fujii, M., … & Horimoto, T. (2020). Detection and characterization of bat Sarbecovirus phylogenetically related to SARS-CoV-2, Japan. Emerging infectious diseases, 26(12), 3025.
Pekar, J., Worobey, M., Moshiri, N., Scheffler, K., & Wertheim, J. O. (2021). Timing the SARS-CoV-2 index case in Hubei province. Science, 372(6540), 412-417.
Pekar, J. et al., (2022) SARS-CoV-2 emergence very likely resulted from at least two zoonotic events.
Rahalkar, M. C., & Bahulikar, R. A. (2020). Lethal pneumonia cases in Mojiang Miners (2012) and the mineshaft could provide important clues to the origin of SARS-CoV-2. Frontiers in public health, 8, 638.
Segreto, R., & Deigin, Y. (2020). The genetic structure of SARS‐CoV‐2 does not rule out a laboratory origin: SARS‐COV‐2 chimeric structure and furin cleavage site might be the result of genetic manipulation. BioEssays, 2000240.
Sirotkin, K., & Sirotkin, D. (2020). Might SARS‐CoV‐2 have arisen via serial passage through an animal host or cell culture? A potential explanation for much of the novel coronavirus’ distinctive genome. BioEssays, 42(10), 2000091.
Speciale, A. C. (2021). Commentary: lethal pneumonia cases in mojiang miners (2012) and the mineshaft could provide important clues to the origin of SARS-CoV-2. Frontiers in Public Health, 869.
Stern, A., Fleishon, S., Kustin, T., Mandelboim, M., Erster, O., Mendelson, E., … & Zuckerman, N. S. (2021). The unique evolutionary dynamics of the SARS-CoV-2 Delta variant. medRxiv.
Temmam, S., Vongphayloth, K., Salazar, E. B., Munier, S., Bonomi, M., Regnault, B., … & Eloit, M. (2022). Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature, 1-10.
Truong, T. T., Ryutov, A., Pandey, U., Yee, R., Goldberg, L., Bhojwani, D., … & Bard, J. D. (2021). Persistent SARS-CoV-2 infection and increasing viral variants in children and young adults with impaired humoral immunity. MedRxiv.
Wacharapluesadee, S., Tan, C. W., Maneeorn, P., Duengkae, P., Zhu, F., Joyjinda, Y., … & Wang, L. F. (2021). Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nature communications, 12(1), 1-9.
Zhou, H., Chen, X., Hu, T., Li, J., Song, H., Liu, Y., … & Shi, W. (2020). A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Current Biology, 30(11), 2196-2203.
Zhou, H., Ji, J., Chen, X., Bi, Y., Li, J., Hu, T., … & Shi, W. (2021). Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. BioRxiv. [Now published in Cell: Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses: Cell]
Zhou, P., Yang, X. L., Wang, X. G., Hu, B., Zhang, L., Zhang, W., … & Shi, Z. L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579(7798), 270-273.
If this article was useful to you please consider sharing it with your networks.

Hi Jonathan,
I like your new paper. How do you remain so calm? I like “For all his phylogenetic maneuvering and aggressive language, he fired only blanks and his targets were straw men and red herrings.” – an excellent sentence I think you have them “over a barrel”.
Has anyone looked in detail at the long branches leading to variants? If those branches are evidence of when the virus was hidden in an immunocompromised person then one might expect a change in the evolutionary parameters – change. In GC ratio, etc – which would then return fast to normal afterwards. Secondly I haven’t had time to read up w hat is known about the involvement of recombination in normal coronavirus replication, but I expect there are some clues there. Anyway, congrats. Keep up the great efforts. Best wishes. Adrian
PS I have decided that there is a new area called ‘political virology’ that produces papers with lots of authors – the greater the number, the more the politics!
Hi Adrian
Great questions.
Re examining the long branches: From the stern paper (The unique evolutionary dynamics of the SARS-CoV-2 Delta variant): “Most variants are characterized by long branches leading to their emergence, with an excess of non-synonymous substitutions occurring particularly in the Spike and Nucleocapsid proteins”. This excess implies a strong selection pressure.
Re recombination: the acceleration of evolution in variants occurring in immunocompromised/vulnerable patients we think is due to recombination in combination with high levels of variation in those individuals. In normal coronavirus evolution recombination occurs but since genetic variation is normally very low (due to bottlenecks: because when people acquire the virus they receive just a few near-identical viruses and have little time to acquire mutations) its’ effects are not readily apparent. When variation builds up in single patients harbouring the virus for a long time, then recombination becomes super important because it allows mutations in separate genomes to recombine with each other and so create variants with multiple mutations. Hence variation accumulates at a far greater rate, which equates to more rapid evolution. It is important to factor in also that many virus mutations only provide a benefit when they are combined, i.e. they are epistatic. As you will know epistasis is when there are synergies between mutations, but normal epistasis requires the mutations to be present in the same genome, which again is facilitated by recombination and so difficult to achieve in normal transmission. The closest (but still inadequate) peer-reviewed discussion of this is in the Hill et al., 2022 paper, found in the reference list. This all applies in spades to the miners because the selection pressure was greater and they were quite likely infected by multiple viruses.
Jonathan
So we are to believe that WIV let the PhD student use a faulty assay before it was validated? What prompted them to figure out the test was a false positive? There must have been some kind of process and other assays involved, yet there is no explanation about this. Additionally, that means the samples DID test positive at their lab even if they were false positives. Why was that omitted from the addendum?
It is important to know the ful list of the participants of the Fauci tel conference on 1-2-2020.
Can you mention their names?
Good suggestion that I should have done in the first place.
From the Feb 1st agenda email sent by Jeremy Farrar:
“Kristian Anderson
Bob Garry- I have not been able to contact Bob. Please forward if you can.
Christian Drosten
Tony Fauci
Mike Ferguson
Ron Fouchier
Eddie Holmes
Marion Koopmans
Stefan Pohlmann
Andrew Rambaut
Paul Schreier
Patrick Vallance”
From: https://s3.documentcloud.org/documents/20793561/leopold-nih-foia-anthony-fauci-emails.pdf
p3134
Jonathan Maybe you can write an exciting story about the Swat team and the Raccoon dog?
thanks ,you did on 29-june 2023