
By Jonathan Latham, PhD and Allison Wilson, PhD
This text was originally posted as a Twitter/X thread. We are reposting it (minus a few typos) as a short essay.
1/ Many people on Twitter are, like Nicholas Wade, wondering if the latest DEFUSE grant revelations from @emilyakopp and @USRTK are maybe the last word on the great #covidorigin #lableak debate. They are not.
2/ Why? Because they only deepen the mystery. The short answer is that six hallmark sites of reverse engineering identified by Bruttel et al: (https://www.biorxiv.org/content/10.1101/2022.10.18.512756v1) only explain 0.1% or less of the #SARS-CoV-2 genome.
3/ If we provisionally accept they demonstrate a #lableak, and the evidence is clear we should, where nevertheless did the rest of the genome come from? Might that not be interesting to know?
4/ It well might, because even though SARS-CoV-2 shows evidence of manipulation, it ALSO shows equally clear evidence of evolution and selection for a human host. A compelling contradiction!
5/ The most recent evidence for that evolution can be found in a paper from July ’23 (Ou et al, 2023) that got few reads, hardly any citations, and practically no interest on Twitter so far as we can tell. We found it only last week. Probably, it was ignored in part because it’s from China.
6/ Now China surely has scientific censors and so credibility needs to be earned but most of Ou et al’s experiments can be corroborated by evidence generated outside China. Here is a link to the Ou et al paper: https://www.nature.com/articles/s41421-023-00581-9
7/ It evaded the censors probably because in China they presume this work proves an evolutionary origin and so refutes a #lableak. This is a logical mis-step for which we should nevertheless be grateful. Here are the findings. It’s a big paper with multiple key conclusions.
9/ (Btw the researchers are from prestigious institutions in Beijing, the English is excellent and the rationales and logic are mostly crystal clear). So strap in tight! PLUS, we are listing the findings in order of increasing magnitude, so try to read to the end.
10/ The basic research idea is to look at the closest spike relatives of SARS-CoV-2 (Specifically BANAL-52 and BANAL-236, found by French/Laotian researchers in N. Laos) and compare them for structure and function, and sometimes including SARS1.
11/ They focussed specifically on 1) receptor (ACE2) binding to humans and bats; 2) Amino Acid (AA) sequences; 3) protein 3D structures; and 4) virus cell entry efficiency. (Henceforth SARS-CoV and SARS-CoV-2 will be called here SARS1 and SARS2.)
12/ There are 3 principal results:
RESULT 1: Binding of SARS1, SARS2, BANAL-52 and BANAL-236 (henceforth B-52 and B-236) to putative host animals.
13/ The BIG findings: Binding of SARS2 spike was strongest to humans. SARS2 binding to Raccoon dog ACE2 was quite weak. After humans, the strongest binding by SARS2 was pangolin and civet cat ACE2s (see Fig. 1.).
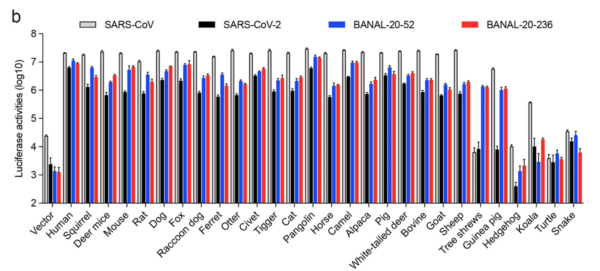
14/ Note too that, with Raccoon dog ACE2 binding, SARS1 and B-52 and B-236 all bind better than SARS2, which trails about 5-fold behind (note the log scale). If anything, a Raccoon dog zoonosis scenario would expect SARS2 to bind BETTER, not less, than the ancestor.
15/ The above experiments need reproducing independently (but by who?) but it’s a bad look for the current zoonosis favourite (and may explain why no Western lab has reported this obvious, key and easy, expt., and one to cite to the “We’ll never know” crowd).
16/ RESULT 2 The authors found that B-52 and B-236 spikes bound well (or very well) to all 14 Rhinolophid bat ACE2s tested. In contrast, SARS2 bound less strongly than B-52 and B-236 to all and very weakly to some bat ACE2s. SARS2 ALWAYS bound less than either B-52 or B-236 (See Fig 2.).
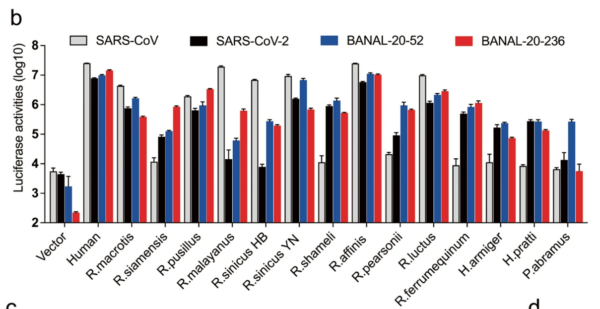
17/ This weak binding by SARS2 to bat ACE2s is mainly due to a single AA difference: H498Q . SARS2 has a Q (Glutamine) at position 498 while the BANALs carry an H (Histidine).
18/Their evidence for this:
B-52 and SARS2’s entire spikes differ by just 19 amino acids (AAs) and only at that one position 498 in the RBM domain (the bit that binds to ACE2).
19/ The authors tested the effect of this substitution. When SARS2 was changed to H it bound MUCH BETTER to some bat ACE2s (not all seem to have been tested). Conversely, changing H498 to Q in B-52 gave MUCH REDUCED binding to the same bat ACE2s (see Fig 3.).
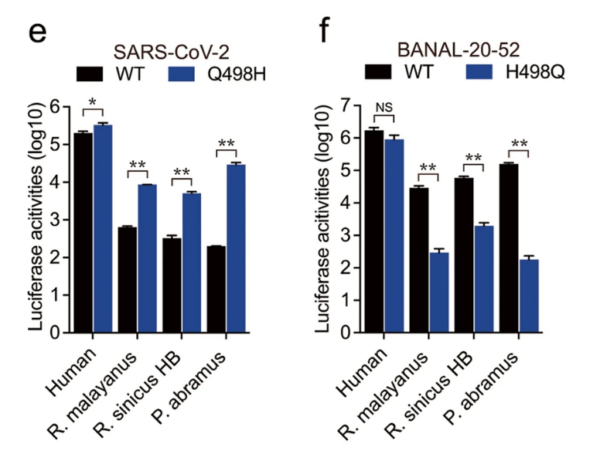
20/ Potentially, this change was simple happenstance that occurred in a host switch since the change DOES NOT affect binding to the human ACE2–and therefore was not a product of selection on that account. But there is more.
21/ The authors also raised antibodies against each spike protein. They then tested the Abs raised against B-52 for their activity against SARS2 BOTH WITH AND WITHOUT the Q498H change. Having a Q made SARS2 2.2-fold less sensitive to Abs (see Fig 4.). This is a lot for a single AA difference.
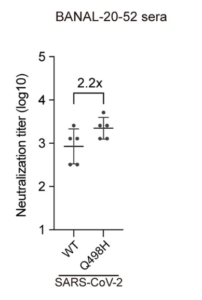
22/Their conclusion was that 498H is highly immunogenic. They estimate the Q on SARS2 lets it evade 25% of the Abs raised against B-52. Maybe just chance. However, this finding offers a reason why SARS2 lost its H (which is conserved in all the RBMs of closely related bat coronaviruses–i.e. those from pangolins and BANAL-103) in a new host.
23/ BUT such an explanation is only relevant in hosts with strong acquired immunity (like longterm patients or immune/vaccinated populations). Not many spillover theories fit that bill. But see below.
24/RESULT 3: Here, the rubber really meets the road: The authors deciphered the 3-D structure of the B-52 and B-236 spikes. In the closed conformation both were very compact compared to SARS2.
25/ Both also had an extra glycan (carbohydrates attached to proteins, often for physical protection) at position 370. This loss of glycan was already known: https://pubs.rsc.org/en/content/articlehtml/2021/sc/d1sc04832e
26/ SARS2 lacks this glycan because of a Threonine to Alanine difference at position 372 (T372A) of the SARS2 spike. (This T forms the third AA in a motif (NXT) needed for glycosylation, thus the glycan attaches to the N at position 370 even tho the mutation is at 372.)
27/ This glycosylation is also highly conserved among betacoronaviruses. Even MERS and SARS1 have it, as do all close relatives. Likely it’s important to them (See Fig. 5.).

28/ Previously, Kang et al had shown that removing this glycan from the spike of SARS2 enhances its affinity for the human ACE2 receptor by 20-fold. That’s big! Kang: https://www.sciencedirect.com/science/article/pii/S0092867421008333
29/ Why do all these bat viruses forego that degree of receptor binding enhancement? What does T372 do for the bat viruses that makes it worth keeping?
30/ Recall that bat coronaviruses are ALL pathogens of the GI tract and that the B-52 and B-236 spikes were compact. It turns out that mimicking the bat small intestine (with pH 5.5 and adding the protease trypsin) causes A372 mutants of B-52 and B-372 to disintegrate.
31/ In sum, the 370 glycan protects the spike, which is on the surface of the virus, from degradation in the bat gut, but this physical protection is at the cost of reducing spike receptor binding. The specific mechanism is probably that the open position of the spike (the “up” position), which is required for receptor binding, also exposes it to digestion.
32/ Hence the T or A choice at position 372 represents a trade-off, and in the lung, where proteases are much less abundant and the pH is more neutral, A is favoured over T. Hence glycan 370 isn’t needed in lungs and the virus can take advantage of a more open conformation.
33/ Thus SARS2 is adapted to lungs. I checked, and it takes two nucleotide changes for the ancestral T in any of the known ancestors to become an A in SARS2. Thus two successive mutations need to occur next to each other. That takes quite a bit of evolution.
34/ Of the two mutations, one is synonymous (does not change the AA) and the other is non-synonymous (it changes the AA). In a population of viruses, non-synonymous mutations can change frequency very rapidly but synonymous mutations typically change frequency only slowly.
35/ Ou et al offer very limited clues as to what they think; But here is what we think:
1) The Raccoon dog result (RESULT 1) is intriguing as yet one more reasonable but inconclusive argument against a Raccoon dog zoonosis.
36) 2) The clear implication of RESULT 3* is that the Glycan 370 mutation did not happen in bats (where it would be highly deleterious). It could have mutated in an intermediate host but it would have taken a considerable time.
37/ Extended time in a zoonosis implies a substantial but unnoticed infection chain in intermediate hosts. It happened with SARS1 but it left evidence.
38/ A lot of infected animals ought to be apparent, either 1) from their sickness, OR, 2) through multiple spillovers. Recall that SARS1 spread in animals and crossed to humans multiple times. Likewise MERS. However, in this case there is evidence only for A SINGLE spillover.
39/ Recall also that the SARS2 spike LOST affinity for Raccoon dog spike compared to its BANAL ancestors. If these were the intermediate host and yet the spike spent an extended period of time in these animals, why would it lose much of its affinity for their ACE2?
40/ RESULT 3, however, is consistent with our Mojiang Miner theory. For people not familiar it is described here https://www.independentsciencenews.org/commentaries/a-proposed-origin-for-sars-cov-2-and-the-covid-19-pandemic/
41/ In a nutshell, we proposed that SARS2 evolved in one of six miners who contracted a mystery COVID-like disease in 2012. We theorised that, just like omicron, the bat coronavirus they probably had, evolved inside them given their long-term hospitalisations.
42/ The WIV (we know) took multiple samples from them. The WIV has since denied these samples contained a coronavirus, but this is disputed: https://www.independentsciencenews.org/commentaries/a-chinese-phd-thesis-sheds-important-new-light-on-the-origin-of-the-covid-19-coronavirus/
43/ What is especially interesting to us is that evolution of a bat virus into SARS2 inside a single human host is FULLY COMPATIBLE WITH RESULTS 1, 2, AND 3. The miner theory offers what a #Zoonosis likely cannot: evolution outside a bat and in the presence of acquired immunity.
44/ The implications for a #lableak scenario are a bit different. Genetic engineering alone did not make SARS2. This was not a simple case of reverse engineering a bat backbone or even adding a furin site to it. Too much AND too many steps are required.
45/ If multiple mutations in addition to a furin site were necessary for SARS2 to infect human lungs as well as it does, maybe subsequent passaging and selection in cell culture or animal models supplied the difference?
46/ There are precedents for passaging engineered coronaviruses https://link.springer.com/article/10.1186/1743-422X-8-64
However, they are considerably less sophisticated than would be required in this case. This would seem to require immune competence, ACE2 humanisation, lungs, and a long time period.
47/ We predict, therefore, that 2024 might be a good year for the Mojiang Miner Evolution theory (#MMPtheory). It is the easy (parsimonious) way to reconcile all these data points, plus all the evidence that is already out there, and maybe the only way.
48/ First up, I will be presenting a poster about it at the upcoming Society of Virology Conference in Vienna, starting March 25th. I’ll be at Poster #429 on Monday 25th March. #GfV2024
49/ It should be interesting, especially given the tumult last time last time I went to a virology conference:
*Footnote: Yuri Deigin pointed out to us, (via Twitter) that the DEFUSE grant application has a discussion of altering glycans on the spikes of the bat coronaviruses they find. The SARS2 spike has 22 glycans. The DEFUSE grant discussion is somewhat vague; it didn’t mention the glycan at N370 specifically, and nor did the papers they cite, and the proposed alterations were for other reasons since the significance of the 370 glycan is new information, but its glycan discussion raises the possibility that this too might have been manipulated.
If this article was useful to you please consider sharing it with your networks.

#34 is, I think, back to front. Otherwise great ideas. Cheers. Adrian
Here is the tweet:
“34/ Of the two mutations, one is synonymous (does not change the AA) and the other is non-synonymous (it changes the AA). In a population of viruses, non-synonymous mutations can change frequency very rapidly but synonymous mutations typically change frequency only slowly.”
It was meant that way. Genetic drift is usually slow but selection can be very fast. Adrian is correct. It can be the other way round but which one it is depends on the selection pressure. Selection typically preserves and conserves amino acids and lets silent mutations drift and that is presumably what Adrian is thinking of. However, in this case, we are suggesting selection pressure for the change would have been strong. In such cases silent mutations can hitch-hike if they are linked to a mutation that is under selection. But the silent mutation has to have occurred in the first place.
wow
It seems a bit strange that another lethal coronavirus outbreak (MERS) would happen less than 10 years after the original SARS outbreak in 2003. Not that I’m into conspiracy theories, but what are the odds?
New details of how the Chinese government has suppressed efforts to determine the origin of SARS-CoV-2 are reported in “Toxic: How the search for the origins of COVID-19 turned politically poisonous,” at
apnews.com/article/china-covid-virus-origins-pandemic-lab-leak-bed5ab50dca8e318ab00f60b5911da0c
The Associated Press reporters write from Beijing.
This article is rather disingenuous IMO. No reference to the US covering things up, just China. Also notice this:
“Some people both inside and outside China speculated about a laboratory leak. Those suspicious included right-wing American politicians, but also researchers close to WHO.” No link to those researchers at the WHO. I don’t know who they would be.
“No reference to the US covering things up, just China.” Of course, it is the Assoc. Press! Get half a loaf from them, find the other half elsewhere, as in your columns.
In our articles we supply multiple angles and mention caveats etc but we choose the topic carefully for the fact that there is a clear conclusion to be drawn but which ordinarily does not get drawn. Otherwise we don’t write about a topic if there is no clear take-home. In the case of AP they provide a clear take-home when the data is, in fact, ambiguous. There is an important difference. One is a clarification the other is a mystification. Nevertheless, we appreciate you posting this link.
It’s great that some of you scientists have not sold your soul and instead use your expertise to make a stand against moral corruption, making a clear, practically inevitable conclusion. The problem is just in the fact that the clear conclusion for some of you is nonsense for other among you:
https://sanityunleashed.substack.com/p/the-many-holes-in-thelab-leak-hypothesis
What do you think should the non-expert do?
The “No-virus” theory is bunk and, to me, obviously so. The SARS2 virus has been isolated and re-isolated hundreds of times and tracked by PCR/sequencing/phylogenetics millions of times. The fact that some (many?) hospitals ran fake tests and falsified their numbers does not change this. My hunch is that the “No-virus” leaders are paid to write this by some as-yet unidentified (but we could guess) forces that want to divide and discredit the critics. Some of this hunch is due to the fact that one prominent No-virus author agreed to debate with me in public but the terms they set for the debate were ridiculous, so I backed out. E.g. they wanted me to share all the papers I wanted to cite with them first, without being clear where the debate was going to go. And so on. Jonathan