
by Jonathan Latham, PhD and Allison Wilson, PhD
Back in March, the World Health Organisation’s report on the origin of the COVID-19 pandemic coronavirus confirmed something that had long been widely presumed. Since the pandemic began, there has been an enormous virus hunt in China.
The purpose of this hunt has been to find the viruses intermediate between SARS-CoV-2 and its coronavirus relatives found in bats (Luk et al., 2019).
The closest known wild relative of SARS-CoV-2 was found by Zheng-li Shi of the Wuhan Institute of Virology (WIV) in a bat in central Yunnan province, China. This virus, called RaTG13, is 96.1% similar to SARS-CoV-2. This genetic difference (3.9%) corresponds to about 1150 nucleotide differences between the two viruses; i.e. it is quite a large gap. Finding intermediate viruses would solve two puzzles. One is geographical: By what means or in what host animal(s) did the virus get to Wuhan? The second is genetic: what viruses were the evolutionary intermediates between RaTG13 and SARS-CoV-2?
The targets of this hunt have therefore been bats but also potential intermediate host animals, such as civets or mink, either one of which might have been the vector that brought COVID-19 to Wuhan. Even partial evidence for such a trail of viral intermediates would support a likely zoonotic origin of SARS-CoV-2.
To this end, according to that WHO report, scientists across China have sampled and tested over 80,000 animals, including 1,100 bats just in Hubei province, of which Wuhan is the capital. Yet beyond a few tantalising discoveries, which are discussed below, the search has been unsuccessful.
The broad failure of this enormous research effort has been scantly reported by the media and sometimes its significance has been dismissed entirely. Thus, the editor of Nature journal recently told the Times Higher Education Supplement that there was an “absence of new evidence” on the COVID-19 origin question. Only a handful of mass media articles and none in the scientific literature have thus done proper justice to the negative results of the sampling in China. Exceptions are “No one can find the animal that gave people covid-19“in the MIT Technology Review and an excellent article by Rowan Jacobsen in Newsweek that expertly articulated the essential points.
Parallel to the hunt inside China, a broader international one has taken place across neighbouring Asian countries. This hunt has mainly focussed on testing bats, which are the reservoir hosts of most coronaviruses. Unlike most of the Chinese search, its results have been reported in the scientific literature (e.g. Lee et al., 2020). As a consequence, in 2021 alone, a series of very near relatives of SARS-CoV-2 have been published. These derive from Japan (Murakami et al., 2021), Cambodia (Hul et al., 2021), Thailand (Wacharapluesadee et al., 2021), and Yunnan province, China (Zhou et al., 2021; Li L. et al., 2021).
The findings of this international search have likewise been poorly covered by the media; either ignored, or, much more rarely, misrepresented (Lytras et al., 2021).
The purpose of this article is therefore to straighten the record. It shows that the positive and negative results of these unprecedented searches are of profound importance for understanding the origin of SARS-CoV-2.
Since the consequences of the Chinese search are fairly simple and better known, this article focuses mainly on analysing and interpreting the published results of the international virus search.
In this article we reveal that the new coronavirus genomes from Asia contain sufficient information to narrow down the geographical source of the direct bat progenitor of SARS-CoV-2 to a quite small region, the south-central part of the Chinese province of Yunnan. In other words, this analysis identifies with good confidence and quite precisely the location where a bat virus that ultimately became SARS-CoV-2 left its bat reservoir host, initiating the chain of events that led to the COVID-19 pandemic.
The analysis does not specify the precise nature of this initiation event. The jump out of bats may have been into an intermediate host (that later went on to infect a human), or it may have been a jump directly into a human; or even the virus may have been procured as part of a research project.
Nevertheless, such a very substantial narrowing of the location of the jump from bats represents a major step forward. Its implications for understanding the origin of SARS-CoV-2 are profound because the requirement for a Yunnan connection markedly constrains origin theories. For example, advocates of the imported frozen food theory favoured in China now have to explain how imported food came to Wuhan carrying a virus from Yunnan (Zhou and Shi, 2021). Likewise, ideas that have circulated about possible European origins of the virus must now explain how a European patient zero could have acquired that virus from Yunnan. Also importantly, the bioweapon theory of Dr Li-Meng Yan is ruled out by the newly discovered viruses discussed here.
But perhaps the greatest significance of this finding will turn out to be that the region of Yunnan indicated as the likely geographic origin is centred on a place called the Mojiang mine. This mine is already well-known to COVID-19 origins investigators.
The Mojiang mine was the site, in April 2012, of an apparent coronavirus outbreak. This outbreak affected six miners and killed three of them (Rahalkar and Bahulikar, 2020). The miners who became ill were shovelling bat guano, implicating the likelihood of infection by a bat virus. The Mojiang mine is also where RaTG13, the closest known natural relative of SARS-CoV-2 was found by Zheng-li Shi of the WIV. RaTG13 was collected during sampling efforts to determine the cause of the mine outbreak. For these and other reasons, the mine is already the focus of lab origin theories. It is highly suggestive, to say the least, for this new evidence to point so precisely to this location as the source of the SARS-CoV-2 bat progenitor.
The finding is thus rich with irony as well as importance. The Chinese and international searches for SARS-CoV-2-related coronaviruses were supposed to reveal a zoonotic origin and refute a lab leak (Anderson et al., 2020). Instead, they have achieved the almost direct opposite.
Our assessment of the widespread mischaracterisation of all this new evidence–in the media and the scientific literature–is therefore that most scientists and most media still resist evidence when it challenges a zoonotic origin or supports a lab leak. These new results do both.
Conclusion one: Intensive search in China yields no evidence for intermediate hosts
Based on the examples of the previous coronavirus outbreaks, the first SARS (hereafter, SARS One) and MERS, an outbreak trail leading to SARS-CoV-2 ought to begin with a reservoir host, in this case presumably bats (Wang et al., 2006; Corman et al., 2014; Hu et al., 2017; Luk et al., 2019). The virus reached humans because an intermediate animal capable of amplifying the virus (presumably without sickening or dying itself) acquired the virus from bats. This intermediate animal host with its Intermediate viruses should be a species found in close proximity to humans at or near the outbreak site.
Thus, a pool of viruses very highly related (≈99.9% similar) to SARS-CoV-2 should be findable in whatever animal species it was that transmitted the virus to humans. Most likely, these intermediates would be domesticated or farmed or smuggled animals (Opriessnig and Huang, 2020). Thus, in the case of SARS One, Himalayan palm civets used in the restaurant trade were the likely amplifying species; in the case of MERS, domesticated dromedaries were certainly the source (Guan et al., 2003; Azhar et al., 2014).
However, for SARS-CoV-2, no comparable pool of viruses in intermediate hosts has yet been found.
While the pandemic was still young, this absence was unremarkable. But, given the extent of sampling in China, the lack of evidence for any part of a transmission chain from bats in Yunnan to humans in Wuhan now represents a major data point against a zoonotic origin.
This lack is frequently dismissed by comparing how long it took to find the origins of SARS One (2002-4) and MERS (2011-2012). But since those outbreaks a lot of resources have been devoted, in China and elsewhere, to sampling and identifying viruses, particularly coronaviruses (e.g. Latinne et al., 2020). There have consequently been vast improvements in our understanding of virus ecology (for example, we now know about bat reservoirs). At the same time there have been huge cost reductions and major leaps in genome sequencing (especially Next Generation Sequencing), in database technology, in virus taxonomy, and in virus isolation methods.
Consequently, the current failure to find a zoonotic proximal origin profoundly challenges the notion that SARS-CoV-2 has a natural animal source. It is no credit to the media or the scientific community that this finding has received so little attention.
Conclusion two: The international search discovers a SARS-CoV-2 lineage with a pronounced geographical distribution
The second major finding is even more compelling but so far all but completely ignored. It derives primarily from the fruits of the international search for bats infected with coronaviruses.
This international search has yielded viral genome sequences that are close relatives of SARS-CoV-2. All are from various parts of Asia (Hu et al., 2018; Zhou P. et al., 2020; Zhou H. et al., 2020; Hul et al., 2021; Wacharapluesadee et al., 2021; Murakami et al., 2021; Zhou et al., 2021; Li L. et al., 2021). These genomes, found mostly in bats (with a few from pangolins), represent the closest relatives of SARS-CoV-2 known from nature. All are between 79% and 96.1% similar to SARS-CoV-2.
Virtually all of these viruses were unknown before the pandemic began and some are even now published only as scientific preprints. Some are from newly sampled bat populations (e.g. Wacharapluesadee et al., 2021; Zhou et al., 2021). Others come from freezer searches for old untested samples (e.g. Murakami et al., 2021). One is even derived from a reanalysis of previously ignored sequence information from historical samples (Li L. et al., 2021).
These twelve known closest relatives of SARS-CoV-2 are listed in Table 1 below. In date order of publication, Table 1 specifies their viral names, their country or province of origin, the genetic similarity of their whole genomes to SARS-CoV-2 (in %), the distance of their sampling location from the Mojiang mine and the species they were sampled from.
The Mojiang mine, which is in central Yunnan, was selected as the centre for this analysis because it is the location where the nearest naturally occurring relative of SARS-CoV-2, RaTG13, was found, in 2013 by Zheng-li Shi (Zhou P. et al., 2020). The coordinates for the Mojiang mine used here (N 23°10’36 E 101°21’28”) are from Canping Huang’s 2016 PhD thesis since those supplied by Zheng-li Shi (N 23°3’27073″, E 101°37’16074″) in Table S1 of Guo et al., 2021 are clearly incorrect.
It should also be noted that, for the purposes of this analysis, the viruses called YN04/05/08 are treated here as one single virus. This consolidation is merited because they are virtually identical in genome sequence and were found at the same location (Zhou et al., 2021). The same applies to the viruses ShSTT200 and ShSTT182 which are referred to here just as ShSTT200 (Hul et al., 2021).
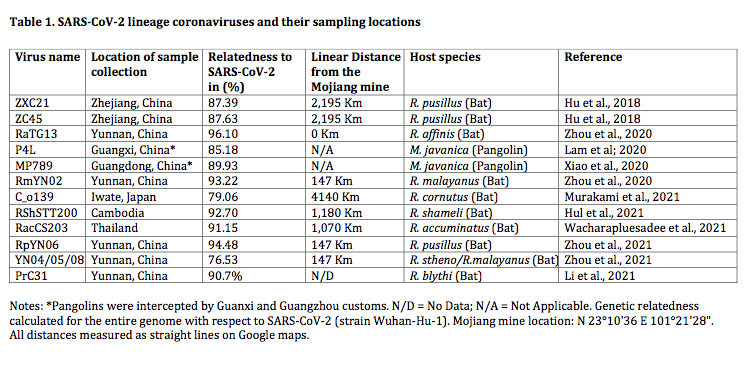
Thanks mainly to these newfound genome sequences, it is now evident that SARS-CoV-2, the pandemic-associated human virus, is just one member of a larger evolutionary lineage. This is seen in the phylogenetic tree shown in Figure 1 below. This lineage has been called the SARS-CoV-2-related lineage (and independently the ‘nCoV’ lineage by Lytras et al., 2021) (Guo et al., 2021).
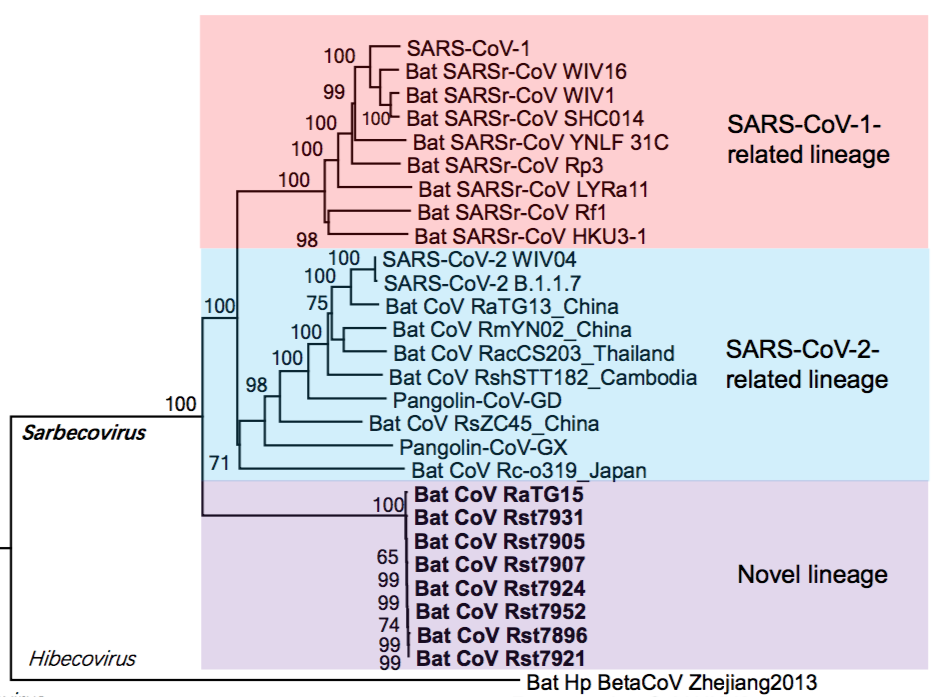
Thus, as shown in figure 1, within the Sarbecoviruses are three lineages. SARS One and its near relatives are at the top (highlighted in pink). At the bottom is a novel lineage (containing RaTG15) very recently reported in a preprint by Guo et al., 2021. In the middle, highlighted in blue, is the SARS-CoV-2 lineage that is the focus of this analysis.
The implication of the existence of all such phylogenetic lineages is that the viruses within them have (for unknown reasons) recombined more-or-less readily with each other, but mostly not with viruses from other lineages (Boni et al., 2020). Otherwise, the lineages would have merged. (We write ‘mostly’ because PrC31, ZXC21 and ZC45 are partial exceptions to this rule, having segments derived from other lineages.) Thus, members of the SARS-CoV-2 lineage are reproductively (i.e., genetically) isolated from the other two lineages. This understanding is key to the analysis below because it means the SARS-CoV-2 lineage can be treated as a distinct group whose members are evolving independently of the other lineages.
By treating this lineage separately, the sampling location and sequence of each virus can be analysed to answer a question that is crucial to the origin mystery. Where in the world did SARS-CoV-2 come from?
In an interview given just after returning from their famous trip to Wuhan, Peter Ben Embarek, leader of the WHO origins investigation team, expressed the following thought to an interviewer:
“[H]aving found other relatively close virus strains to SARS-CoV-2 in the region also in South East Asia where these bats live is a strong indication that’s where the source is”
South East Asia is big place. But Ben Embarek’s statement suggests how one can logically narrow down the possible origins of SARS-CoV-2.
In fact, a more precise analysis than this had already been published. A collaboration between the Wuhan Institute of Virology (WIV) and the EcoHealth Alliance used hundreds of partial viral sequences from China, most of them new to science, to map the geographical origin of SARS-CoV-2 more precisely (Latinne et al., 2020). The authors concluded:
“[W]e found that SARS-CoV-2 is likely derived from a clade of viruses originating in horseshoe bats (Rhinolophus spp.). The geographic location of this origin appears to be Yunnan province” (Latinne et al., 2020) [note: a clade equates here to a lineage].
Relatively little attention was paid at the time to this conclusion. This is largely because the authors provided two substantial caveats. The first was that viruses from outside China were not included in their study. The second caveat was that their analysis used only a small fragment (440 nucleotides) of the virus genome (for most of their samples this was the only sequence information available). A complete coronavirus genome is approximately 30,000 nucleotides. Because recombination between coronaviruses is generally frequent, analysis of complete genomes might reasonably be expected to give different results.
However, due to the new virus discoveries (listed in Table 1), these caveats no longer apply. For the SARS-CoV-2 lineage one can therefore re-do the analysis using complete genomes for all currently identified viruses in the SARS-CoV-2 lineage for which precise geographic location data is available.
None of the researchers who published the novel SARS-CoV-2 lineage viruses in Table 1 performed such an analysis (nor did Lytras et al., 2021, who recently reviewed the evolutionary relationships of the lineage).
However, such an analysis is simple to do. First, though, it requires excluding viruses whose sampling location is uncertain. Hence, those virus sequences extracted from smuggled pangolins (P4L and MP789) are not included in this geographic analysis. This is because a virus found in a pangolin smuggled into China might have originated from almost anywhere in SE Asia.
The other provenance question relates to PrC31. According to the preprint describing it, PrC31 is from “Yunnan” (Li L.et al., 2021). We asked the authors for a more precise location but did not obtain one:
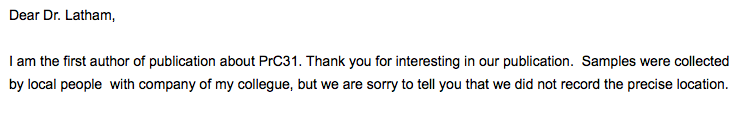
However, according to the NGDC genome database, the accession called PrC31 is from Pu’er City. This matches the initials (which are not explained in the article). Pu’er City is a town 56 km (in a straight line) from the Mojiang mine. Pu’er city, however, is also the name of an administrative district that encompasses the mine. The furthest boundary of this district from the Mojiang mine is 250 km. Thus 250 km marks the maximum and 0 km the minimum presumed distance to the sampling site of PrC31. Given this uncertainty we decided to omit PrC31 from the distance plot (Figure 2 below). However, PrC31 is important since, over certain parts of its genome, it is the closest known virus to SARS-CoV-2. It will therefore be discussed below, where appropriate, as will the pangolin genomes.
Zeroing in
After excluding these viruses, the results are simple to interpret. Table 1 allows a comparison of the degree of relatedness of each virus to SARS-CoV-2 and the sampling location for each virus. The closest relative of SARS-CoV-2 (RaTG13, 96.1% similar at the nucleotide level) was found at the Mojiang mine in Yunnan Province. The next closest genetic relatives of SARS-CoV-2 are RmYN02 (93.2% similar) and RpYN06 (94.48% similar). These two viruses were both also found in Yunnan, just 150 km away (in a straight line) from RaTG13. The next two closest relatives of SARS-CoV-2 are, almost equally, RshSTT200 (92.70%) and RacCS203 (91.15%). These two viruses were discovered 1,180 km away and 1,070 km away, respectively. The next most distantly related (after PrC31 which cannot be pinpointed) are ZXC21 (87.39%) and ZC45 (87.63%). These were found 2,195 km away, followed by C_o319 (79.06%) from Iwate, Japan, 4,140 km away.
There is an obvious pattern here, which is even more evident when Table 1 (minus PrC31 and the pangolin viruses) is plotted out, as in Figure 2.

Thus, with the sole exception of YN04/05/08, every virus in the SARS-CoV-2 clade falls on an almost perfect straight line. Beginning from the discovery location of RaTG13, the further away from the mine a virus was found, the less closely related to SARS-CoV-2 it is.
Thus, if we knew nothing else about the origin of SARS-CoV-2 we would learn from this plot that, first, genetic variation among the bat viruses in this lineage is highly correlated with geographic location.
Second, that the direct bat progenitor of SARS-CoV-2 came from a bat living at or near to the Mojiang mine in south-central Yunnan, China. In other words, the Mojiang area of Yunnan was the site of the key zoonotic leap where SARS-CoV-2’s ancestor exited its bat reservoir. This leap may have been directly into a human. Alternatively, the leap may have been into an intermediate host. The third possibility is that the leap was assisted by scientists collecting or researching bat viruses.
These findings can also be displayed in map form. Figure 3. shows the sampling location of all the viruses plotted in Figure 2.
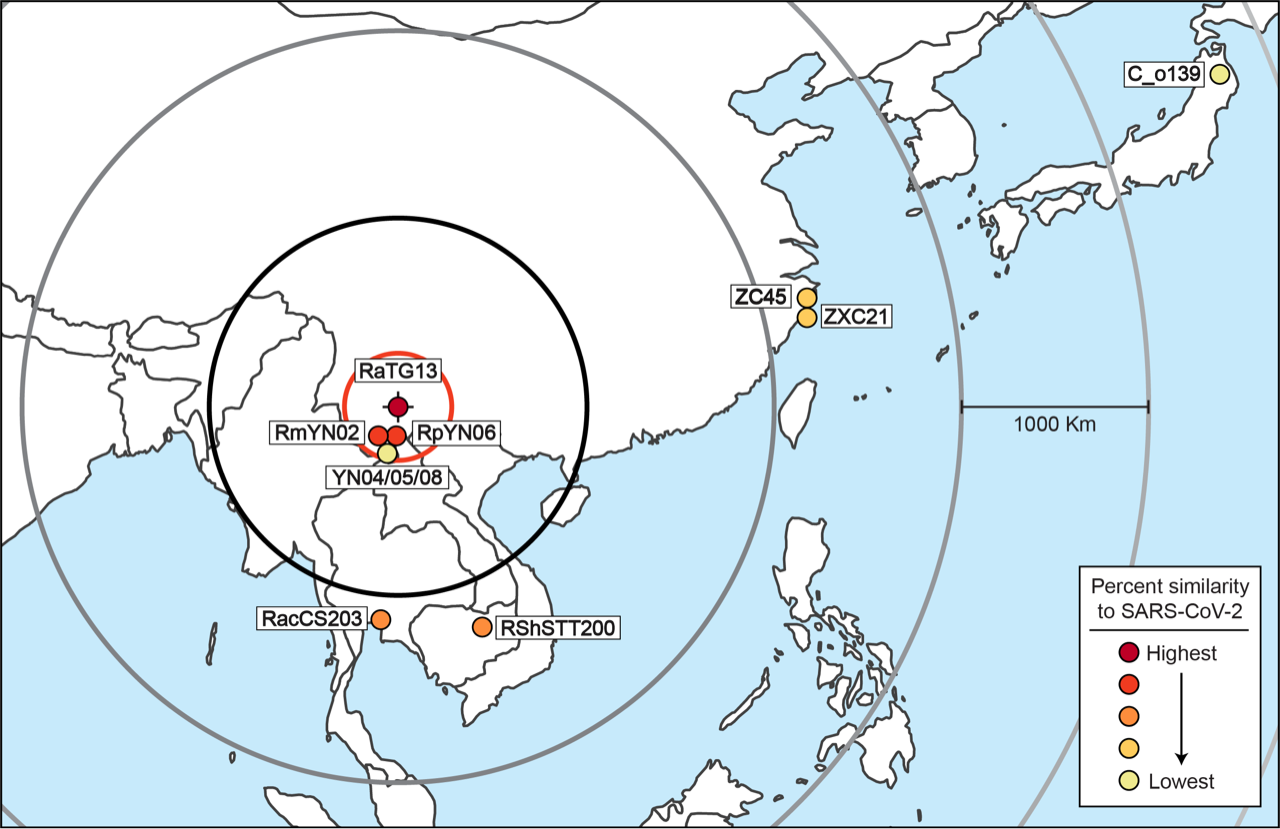
The only outlier in this analysis is YN04/05/08. Its presence in Yunnan can presumably be explained as a less related virus that migrated back towards Yunnan. An alternative possibility is that YN04/05/08 is not recombining with the other viruses in the lineage and is in the process of forming a new lineage. This exception does not refute the overall analysis. Only the discovery of a natural virus that was closely related to SARS-CoV-2 but that was found far away would do that; such a virus would indicate that the progenitor of SARS-CoV-2 might also have originated far from Mojiang. To date, no such virus has been found.
The geography of SARS-like coronaviruses
Combining genome sequences with map locations is an established practice known as phylogeography and there are strong precedents (in addition to Latinne et al, 2020) for studying bat coronaviruses using this methodology.
An important example, which is highly relevant since it also involves SARS-related coronaviruses with very similar bat hosts, is a study titled “Geographical structure of bat SARS-related coronaviruses” (Yu et al., 2019). This was research done by Yu Ping, a student of Zheng-li Shi’s. These authors concluded that viruses in the SARS One lineage circulated freely among the Rhinolophid (horseshoe) bats that are their reservoir hosts (Bannerjee et al., 2019). This lack of host restriction meant that:
“[S]pace presents a greater barrier to virus diversification than host species for the evolution of bat SARSr-CoVs.”
In other words, geographic proximity better predicted the occurrence of specific isolates than did bat host species. So whereas one might have predicted that these viruses moved freely within each species of horseshoe bat and only sometimes switched between them, and thus viral genetic variation would closely track bat species distributions, it seems instead that this lineage of coronaviruses easily switched between the different species of horseshoe bats that are their hosts.
Largely unfettered movement between hosts means that, whenever new virus variants arise or new recombinant genomes arise, these can easily spread within one cave or one roosting site to other species (of horseshoe bat). They have more difficulty disseminating to other caves and sites. Presumably, their bat hosts have life histories or specific behaviors, such as flight path routines or infrequent switching of roosting sites that can explain this limited viral movement. The relevant consequence of this is that, within a lineage, virus location predicts the degree of similarity to other isolates.
Yu Ping’s finding is consistent with a landmark study of SARS-related coronaviruses published by Zheng-li Shi’s lab at around the same time (Hu et al., 2017). While at first sight these findings seem to contradict the applicability of phylogeographic approaches for these viruses, it turns out they are more likely to be the exception that proves the rule.
In 2017 Zheng-li Shi’s group reported finding, in one single location, multiple strains of SARS-related coronaviruses with (between them) the highest known genetic similarity to SARS One, the virus that caused the 2002-04 outbreak (Hu et al., 2017). The site was a cave close to Kunming, capital of Yunnan province.
The authors reached two major conclusions, 1) that the direct bat progenitor of SARS One arose through recombination among precursors of these viruses, 2) that Yunnan was “likely to be the geographical source” of SARS One.
And more broadly:
“SARSr-CoV evolution is strongly correlated with their geographical origin, but not host species.” (Hu et al., 2017)
As the authors acknowledged, this generated what was subsequently termed a ‘mismatch’ (Luk et al., 2019). The puzzle consisted of the fact that the 2002 SARS One outbreak commenced in Guangzhou, Guangdong province (where the virus apparently jumped from civets to humans). Guangzhou is 1,200 km south east of the cave near Kunming where the spillover to humans would have been predicted from the phylogeographic evidence alone.
According to Zheng-li Shi, in comments made at the time to a Chinese online newspaper, this mystery can be resolved:
“The Paper: Is the civet being wronged?
Shi Zhengli: Not wronged. It is a fact that it spreads the SARS virus, it is the intermediate host, and bats are the source.
We went to a township under Kunming, Yunnan. I checked the information at that time. In 2003, there was a civet breeding farm in Kunming, but there is no more now. At that time, the country’s civet cats were sold in Guangdong, mainly for food.” [Google translate]
In other words, Zheng-li Shi had a ready explanation in 2017 (which is not mentioned in Hu et al., 2017) for how SARS One moved from Kunming, Yunnan, to the outbreak epicentre. It likely spread via civets, which have long been considered the likely intermediary host for SARS One (Wang et al., 2006). Presumably, civets being farmed in Kunming became infected via contact with bats. Subsequently, ones infected with the direct progenitor of SARS One were then transported to Guangdong.
The example of SARS One suggests two things. First, that it is indeed practicable and productive to track bat coronavirus reservoirs down to the microgeographical level of a few kilometres. Thus, it would not be surprising, since the SARS One lineage and the SARS-CoV-2 lineage share the same host species (Rhinolophus bats), and these bats rarely fly far afield (Lau et al., 2010), if the SARS-CoV-2 lineage could be similarly tracked.
Second, the successful mapping of SARS One and the strong geographical associations often noted in the virology literature for similar bat coronaviruses (see Latinne at al., 2020 and also Fig. 3 in Boni et al., 2020), make it puzzling that coronavirologists have not already analysed SARS-CoV-2 and its newfound relatives in the same way.
SARS-CoV-2: The provenance of its genome subparts
This analysis has so far established that genetic relatedness among the SARS-CoV-2 lineage of coronaviruses in their bat reservoir is strongly correlated with sampling location. Such a correlation allows viral genome sequence alone to be used to find the geographic source of any bat virus in the lineage if that is not already known. Applied to SARS-CoV-2 this reasoning locates its last bat ancestor to a site at or near the Mojiang mine.
This finding is considerably more than a simple reformulation of the idea that the mine where RaTG13 was found might be important or the conclusion of Latinne et al., 2020, that SARS-CoV-2 might have come from Yunnan.
This phylogeographic analysis greatly strengthens the weight and precision of this association. By showing that the highest related genomes are all nearby and only less related ones far away, the association of the mine with SARS-CoV-2 is not a happenstance but part of a general phylogeographic pattern among the SARS-CoV-2 lineage. This pattern makes it highly probable that the direct bat precursor virus of SARS-CoV-2 came from, at most, within a few hundred kilometres of the Mojiang mine, with the mine itself being the epicentre of the probability gradient, i.e. the most likely single spot.
The approach used above correlated whole genomes with location. A variant of this method is to take into account the fact that different sections of the SARS-CoV-2 genome have independent evolutionary histories due to recombination between viruses (e.g., Boni et al., 2020; Lytras et al., 2021). Dividing up the evolution of the SARS-CoV-2 genome and its related coronaviruses into these independently evolving sections is arguably a more nuanced approach to determining its origin. However, there are trade-offs. Breaking down the genome requires making assumptions about historic recombination breakpoints, and these estimates can introduce errors of their own.
What happens when one does delve down?
If one compares the genome of SARS-CoV-2 with the other members of the SARS-CoV-2 lineage (including PrC31 and the pangolin genomes) by creating a similarity plot (this one generated by Twitter user @Babarlelephant), an important point becomes immediately clear. None of the viruses currently identified can be the sole direct ancestor of SARS-CoV-2, not even RaTG13 (even though RaTG13 is by a considerable way the closest in overall percent similarity).
As the similarity plot shows (by finding the highest line on the plot), some regions of SARS-CoV-2 are clearly genetically closer to RmYN02 (the light blue line) than to RaTG13 (the red line), while for other regions the closest to SARS-CoV-2 is RpYN06 (the black line). Four separate parts in ORF1, meanwhile, are closest to PrC31 (the green line). One very short segment (including the crucial receptor binding domain (RBD) is closest to the Guangdong pangolin genome (MP789) while another very short segment is closest to RacCS203.
The similarity to SARS-CoV-2 shown by these latter two segments, however, should be treated with caution. They are short enough that their apparent close relatedness may have arisen through chance (i.e. they are potential examples of convergent evolution) and not through having a common ancestor.
The key overall point to be learned from the plot is that, for over 99% of the genome of SARS-CoV-2, the closest known genetic sequence is present either in RmYN02, RpYN06, PrC31, or RaTG13. These four viruses are thus the closest relatives of SARS-CoV-2, depending on which part of the genome is examined. This makes SARS-CoV-2 a recombinant whose genome is, effectively, a synthesis of each of these different bat viruses.
Given that these four viruses are all from the same limited region of central-southern Yunnan this is, if anything, a still more convincing demonstration than the whole genome analysis presented above, that this area is the source of SARS-CoV-2.
The spike protein
This discussion has so far taken a simple mathematical approach that omits a crucial aspect of the COVID-19 emergence story–the nature of coronaviral zoonoses.
A zoonotic emergence of a bat coronavirus into humans requires something unusual. Most bat coronaviruses do not infect humans or human cells because they lack a spike protein capable of binding human ACE2 (or, like MERS, another human receptor) (Hu et al., 2017). The spike protein, therefore, as has often been pointed out, has a special role in triggering emergence (Becker et al., 2008; Ge at al., 2013). In fact, in 2014, Zheng-li Shi and Peter Daszak were awarded a US NIH grant to test whether “S(pike) protein sequences predict spillover potential” as measured by their ability to bind human ACE2. Their prediction was that spike binding alone predicts emergence, a suggestion originally proposed by Kuo et al. in 2000.
The inspiration for this approach was their research on the jump into humans of SARS One discussed above. In the cave near Kunming where they found the series of viruses most closely related to SARS One, they also noted that some of these viruses, unusually for bat coronaviruses, had spike proteins that bound human ACE2 (Ge at al., 2013). Experiments were able to show that these particular spikes enabled whatever bat virus carried them to infect human cells (Menachery et al., 2015).
Their working hypothesis became that any bat coronavirus with a human compatible spike could switch species–from bats to humans–regardless of the rest of the genome (Ge et al., 2013). A human-compatible spike was both necessary and sufficient for a zoonotic leap. The cave near Kunming, therefore contained the nearest relatives of SARS One solely because a subset of them had the right spike to unlock human cells using their ACE2 binding ability. Coronaviruses containing this spike then encountered a physical route, via farmed civets, that led to human infections and ultimately the SARS One outbreak (Hu et al., 2017). Thus, once a spike evolved in a bat that could bind the human ACE2, the remaining sequences followed, essentially opportunistically.
The implication for the emergence of SARS-CoV-2 is that, whereas the provenance of each part of the SARS-CoV-2 genome is of equal phylogeographic interest, not all coronavirus genome regions are equal in other ways. The most important region of the genome, so far as zoonotic emergence is concerned, is the part that specifies the spike.
Inspecting the similarity plot again we can see that the closest spike found anywhere is, by a large margin, the one possessed by RaTG13; and RaTG13, we know, was found in the Mojiang mine.
The RaTG13 spike shares 98% amino acid identity with SARS-CoV-2. However, while some researchers have concluded that the spike of RaTG13 binds human ACE2 but only moderately well, others have concluded there is negligible binding (Shang et al., 2020; Wrobel et al., 2020; Li Y. et al., 2020; Li and Zhang 2021; Li P. et al., 2021; Liu et al., 2021; Mou et al., 2021; Guo et al., 2021). But what is much more important than these somewhat inconclusive results is that (unless SARS-CoV-2 was a product of lab enhancement) we can be fairly certain that the progenitor (RaTG13-like) virus which first infected a human, also bound human ACE2, at least to some degree, and that it was this binding that enabled the spillover.
From this premise we can reconstruct a plausible emergence pathway. An RaTG13-like spike, from Mojiang or nearby, led the zoonosis. It combined with genome sequences similar to RmYN02, RpYN06 and PrC31 and these followed in its wake.
Thus, by length of the total genome contributed, RmYN02, RpYN06, RaTG13 and PrC31 were approximately equally important to the rise of SARS-CoV-2. However, from a zoonotic perspective, the spike region contributed by RaTG13 is much the most important. It would have catalysed the outbreak and therefore RaTG13, or some close relation, is the best candidate for being present at the pivotal moment: the infection of patient zero.
The implications for zoonotic theories of an origin in south-central Yunnan
Locating the bat progenitor of SARS-CoV-2 to the Mojiang area of Yunnan has major implications for understanding the origin of SARS-CoV-2.
First, it places substantial constraints on natural zoonotic origin possibilities.
Zoonotic origin theories typically assume a proximal source in farmed or smuggled or wild animals. The analysis developed above implies, however, that any zoonotic theory must plausibly accommodate a bat jump in south-central Yunnan, much as Zheng-li Shi hypothesised for SARS One a little further north.
For example, a widely discussed zoonotic possibility is that SARS-CoV-2 was smuggled or traded into Wuhan, e.g. via “Malayan pangolins illegally imported into Guangdong province” (Anderson et al. 2020; Lam et al., 2020). This pangolin origin possibility is still widely cited, although it has also been the subject of much scientific criticism (Lee et al., 2020; Lytras et al, 2021; Choo et al., 2020; Frutos et al., 2020). The expectation has been that this pangolin reached Wuhan from countries like Malaysia, Cambodia, or Laos, where pangolins are fairly common (Lee et al., 2020). Our phylogeographic analysis indicates, however, that the pangolin must have acquired its virus from the bat reservoir in Yunnan and not in its country of origin or some other part of China. So while acquiring the virus in Yunnan does not rule out a pangolin as a proximal origin or a zoonosis per se, this analysis does constrain these possibilities very significantly.
To choose another example, some apparent very early COVID-19 cases have been reported from Spain, Italy, and France. A Yunnan origin, however, posits that the virus did not ultimately come from Europe.
Thirdly, a south-central Yunnan origin has implications for the suggestion of Chinese scientists that SARS-CoV-2 reached China from abroad via frozen food (Zhou and Shi, 2021).
This idea was apparently taken seriously by WHO investigators but it seems incompatible with a central Yunnan origin. Even if the food came from abroad, the virus contaminating it presumably did not.
Fourth, a zoonosis implies the existence of naturally-occurring intermediate viruses that ought to bridge the genetic gap of 1150 nucleotides between RaTG13 and SARS-CoV-2 (recently estimated at around 40yrs by Lytras et al., 2021 and also Boni et al., 2020). This gap has been partially filled by the discoveries of RmYN02, RpYN06 and PrC31, which in certain genome regions are intermediate in sequence between RaTG13 and SARS-CoV-2. Nevertheless, even taking these viruses into account, about two thirds of the gap in the putative zoonotic trail remains. These hypothetical naturally-occurring intermediates have not been discovered, it is suggested, because bat coronaviruses have been “massively undersampled” (Andersen et al., 2020).
However, a south-central Yunnan origin implies that any undersampling pertains specifically to Yunnan, since this is where all the other close relatives of SARS-CoV-2 have been found.
Is Yunnan undersampled? As we have previously summarised, numerous different virology teams extensively sampled in Yunnan, especially at the Mojiang mine, even before the pandemic struck. For example, Zheng-li Shi’s colleagues alone visited the Mojiang mine seven times in the years following the 2012 outbreak. At least three other teams of virologists sampled the mine looking for coronaviruses prior to the pandemic. By their own accounts, WIV researchers alone took thousands of samples and found hundreds of coronaviruses (Ge et al., 2016; Guo et al., 2016). Post-pandemic, AP documented numerous wildlife sampling research projects in China as part of what it called a “hidden hunt for coronavirus origins” especially in bats, including in Yunnan. Thus, massive undersampling at this point in time seems questionable.
The discussion above demonstrates that pinpointing a specific region of Yunnan as the site of the jump from bats requires zoonotic theories to be more specific and precise in terms of host species, viral intermediates, and their expected locations. This specificity is highly valuable. It should make every theory both easier to confirm or to refute. On the other hand, any theory that cannot be adapted to include a Yunnan origin ought, henceforth, to be considered not credible.
The implications for lab escape theories of an origin in south-central Yunnan
Lab origin theories of SARS-CoV-2 also should have their credibility tested against these new virus sequences. Li-Meng Yan and colleagues have proposed that SARS-CoV-2 is a deliberately released bioweapon. These authors proposed that the backbone of this ‘weapon’ was ZC45 and/or ZXC21. However, because RaTG13, RmYN02, RpYN06 and PrC31 are, depending on the region of the genome selected, invariably closer to SARS-CoV-2 than either of ZC45 or ZXC21, Dr Yan’s formulation of a bioweapon theory can be confidently ruled out.
A Mojiang location constrains other lab origin theories too.
Three distinct categories of lab accident theory have been proposed so far. The simplest scenario is that SARS-CoV-2 resulted from infection of a researcher on a sample collecting trip. This worker could have infected others when they returned to Wuhan. From the present analysis it can be inferred that any such collecting trip would have been to south/central Yunnan. Consequently, it may be possible to effectively rule out this possibility if it could be shown that no virologist from Wuhan travelled to Yunnan province in mid-to-late 2019.
A second category of lab origin postulates that RaTG13 (or a similar virus) was obtained from the Mojiang mine and enhanced or altered for some vaccine or technology-related research purpose. This genetically manipulated or passaged virus then escaped (Kaina, 2020; Segreto and Deigin, 2020; Sirotkin and Sirotkin, 2020; DRASTIC, 2021). Such theories are consistent with any phylogeographic findings since any changes from known viruses can, in principle, be explained by lab manipulation or adapted to propose an alternative source of the viral backbone. Therefore, an origin close to Mojiang is not a major constraint. A much greater one is that these lab origin theories do need to explain why genome sequences resembling the naturally-occurring viruses RmYN02, RpYN06 and PrC31 are found in SARS-CoV-2. Presumably this explanation might be that researchers in Wuhan had access to another virus, one that combined an ORF1 region that was more similar to these sequences with an RaTG13-like spike. This virus was then modified, perhaps by inserting a furin cleavage site. The expectation would nevertheless be that this hypothetical virus came from south-central Yunnan.
The third category of lab escape is our Mojiang Miners Passage theory. This is based on the medical cases of the six miners, mentioned above, who all became sick in 2012 whilst shovelling bat guano at the Mojiang mine (Rahalkar and Bahulikar, 2020).
These six miners all developed Covid-19-like symptoms and were diagnosed at the time with a probable novel coronavirus. The theory proposes that a RaTG13-like coronavirus (or mixture of viruses that later recombined into one) from the mine infected the miners. Some of these miners were ill for almost six months. Our suggestion, therefore, is that the bat virus(es) that infected them evolved (through a passaging-like process) inside their bodies to become human-adapted.
Since it is known that numerous medical samples were taken from the miners and many were sent to the Wuhan Institute of Virology, this virus may have escaped when those medical samples were used for research, perhaps to culture the virus or to manipulate it.
We favour this theory because it explains numerous otherwise puzzling features of SARS-CoV-2. These features are (1) the high improbability of a zoonotic appearance of a SARS-related coronavirus in Wuhan; (2) the apparently pre-adapted nature of the virus to humans (Piplani et al., 2021; van Dorp et al., 2020; Zhan et al, 2020); (3) a miner’s passage predicts a single zoonotic jump to humans [which fits the data on early human sequences (Bloom, 2021)] and which is inconsistent with most viral zoonoses, which typically feature multiple jumps into humans; (4) a miner-derived virus also explains the proclivity of SARS-CoV-2 for human lungs, which is a characteristic that many coronaviruses lack; (5) the theory can also explain the extensive attempts to deny or obscure research occurring at the WIV (see also the Zhou P. et al., 2020a addendum). The Mojiang miners hypothesis even has an evolutionary explanation for the infamous furin cleavage site. However, none of this precludes the possibility that the miner-derived virus was also lab-altered.
Since the theory specifically postulates that patient zero was a Mojiang miner who acquired one or more SARS-CoV-2-related viruses directly from the bats in the mine, the miners passaging theory matches perfectly the phylogeography of SARS-CoV-2 lineage revealed above. Indeed, it is an explicit prediction of the Mojiang miner passage theory that SARS-CoV-2 is composed of viruses originating there. Consequently, a miner passage origin is also consistent with SARS-CoV-2 being a mosaic of RmYN02, RpYN06, PrC31 and RaTG13 since, as the phylogeography shows, these viruses, or their close relatives, could have been present in the mine when the miners fell ill.
A miner passage is therefore not just compatible with but greatly strengthened by all the new evidence from wild viruses that has emerged since the pandemic began.
A phylogeographic approach to the SARS-CoV-2 lineage thus provides a striking result on several fronts. Lab origin theories can readily account for a south/central Yunnan origin, since the Mojiang mine is already their starting point. But while the various lab leak theories have their differing explanations (evolution in the miners/genetic engineering/lab passaging) for how RaTG13 (or similar viruses) might have given rise to SARS-CoV-2, a natural zoonotic origin relies on evolution in wild (or at least semi-natural) settings and this should leave traces in the form of intermediate viruses. It is therefore a highly problematic state of affairs for all zoonotic theories that, 1) no viruses with an overall similarity higher than RaTG13 have been found and, 2) that no intermediate viruses from potential intermediate hosts have been found. We can now conclude, however, that Yunnan is the place where such searches should have succeeded.
To sample or not to sample
If a bona fide closer relative of SARS-CoV-2 were found tomorrow in a bat far away from south-central Yunnan, then the genetic distribution of SARS-CoV-2 progenitors would have to be rethought and the special significance of south-central Yunnan would stand refuted.
One obvious approach is therefore to call for more sampling to test the association. Yunnan would be the logical focal point of this search.
However, there is a clear problem with further sampling. It is likely that the SARS One pandemic originated from a bat virus from Yunnan that had evolved the ability to infect humans. The 2012 miner outbreak likewise exemplified the risks of close contact with bat coronaviruses. Furthermore, the phylogeographic analysis presented here greatly strengthens the case, already strong, that SARS-CoV-2 ultimately resulted from virus sampling.
So the paradox is rather acute. What or who will ensure that future sampling is conducted with far greater prudence than virologists have so far mustered?
There is one further crucial issue. To date, both the Wuhan Institute of Virology and the EcoHealth Alliance (EHA) in New York have refused requests by Congress and others, to allow public access to their existing coronavirus samples and their viral databases. These may hold answers to all the origin questions. But if publicly-funded virologists will not share the samples they already have, and are apparently unwilling to face the conclusions public access might entail, why should anyone reward them to collect more? Indeed, how can research into the origins of COVID-19 meaningfully proceed if virologists will neither share their data nor follow where it leads when they do?
The abject failure of the WHO and also of established science, in China and elsewhere, to genuinely investigate the origin question can thus be explained. The problem is not lack of data. As this article and the creative approaches of members of DRASTIC, and others, have shown, there is plenty of valuable data waiting to be brought forth. Rather, the obstacle is simply a deep and broad fear on the part of the scientific establishment that the trail might lead to a lab leak.
The lack of outrage, or even concern, among the rank and file of the scientific community at the flagrant obstructionism of the WIV and the EHA demonstrates the extent of this fear as clearly as could be wished.
The underlying problem is that academic science is enmeshed in a wider transnational Pandemic Virus Industrial Complex (PVIC) that has sought to suppress lab origin theories and within which the WIV and the EHA are just minor cogs.
The important consequence of this is that outbreak origin investigations are always challenging. They require people who are expert but are either not conflicted or who have demonstrated their independence. Consequently, the best data and analysis on the origin of SARS-CoV-2 will continue to come, we predict, mainly from individuals acting independently of established institutions.
Acknowledgements: the authors are deeply grateful to Francisco de Asis, @Babarlelephant, and the other reviewers of this article for their generous assistance and numerous helpful suggestions.
References
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature medicine, 26(4), 450-452.
Azhar, E. I., El-Kafrawy, S. A., Farraj, S. A., Hassan, A. M., Al-Saeed, M. S., Hashem, A. M., & Madani, T. A. (2014). Evidence for camel-to-human transmission of MERS coronavirus. New England Journal of Medicine, 370(26), 2499-2505.
Banerjee, A., Kulcsar, K., Misra, V., Frieman, M., & Mossman, K. (2019). Bats and coronaviruses. Viruses, 11(1), 41.
Becker, M. M., Graham, R. L., Donaldson, E. F., Rockx, B., Sims, A. C., Sheahan, T., … & Denison, M. R. (2008). Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proceedings of the National Academy of Sciences, 105(50), 19944-19949.
Bloom, J. D. (2021). Recovery of deleted deep sequencing data sheds more light on the early Wuhan SARS-CoV-2 epidemic. bioRxiv.
Boni, M. F., Lemey, P., Jiang, X., Lam, T. T. Y., Perry, B. W., Castoe, T. A., … & Robertson, D. L. (2020). Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nature microbiology, 5(11), 1408-1417.
Choo, S. W., Zhou, J., Tian, X., Zhang, S., Qiang, S., O’Brien, S. J., … & Sitam, F. T. (2020). Are pangolins scapegoats of the COVID‐19 outbreak‐CoV transmission and pathology evidence?. Conservation Letters, 13(6), e12754.
Corman, V. M., Ithete, N. L., Richards, L. R., Schoeman, M. C., Preiser, W., Drosten, C., & Drexler, J. F. (2014). Rooting the phylogenetic tree of middle East respiratory syndrome coronavirus by characterization of a conspecific virus from an African bat. Journal of virology, 88(19), 11297.
Segreto, R., & Deigin, Y. (2021). The genetic structure of SARS‐CoV‐2 does not rule out a laboratory origin: SARS‐COV‐2 chimeric structure and furin cleavage site might be the result of genetic manipulation. BioEssays, 43(3), 2000240.
Frutos, R., Serra-Cobo, J., Chen, T., & Devaux, C. A. (2020). COVID-19: Time to exonerate the pangolin from the transmission of SARS-CoV-2 to humans. Infection, genetics and evolution, 84, 104493.
Guan, Y., Zheng, B. J., He, Y. Q., Liu, X. L., Zhuang, Z. X., Cheung, C. L., … & Poon, L. L. M. (2003). Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science, 302(5643), 276-278.
Ge, X. Y., Li, J. L., Yang, X. L., Chmura, A. A., Zhu, G., Epstein, J. H., … & Shi, Z. L. (2013). Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature, 503(7477), 535-538.
Ge, X. Y., Wang, N., Zhang, W., Hu, B., Li, B., Zhang, Y. Z., … & Shi, Z. L. (2016). Coexistence of multiple coronaviruses in several bat colonies in an abandoned mineshaft. Virologica Sinica, 31(1), 31-40.
Guo, H., Hu, B., Si, H. R., Zhu, Y., Zhang, W., Li, B., … & Shi, Z. (2021). Identification of a novel lineage bat SARS-related coronaviruses that use bat ACE2 receptor. bioRxiv.
Hu, B., Zeng, L. P., Yang, X. L., Ge, X. Y., Zhang, W., Li, B., … & Shi, Z. L. (2017). Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS pathogens, 13(11), e1006698.
Hul, V., Delaune, D., Karlsson, E. A., Hassanin, A., Tey, P. O., Baidaliuk, A., … & Duong, V. (2021). A novel SARS-CoV-2 related coronavirus in bats from Cambodia. BioRxiv.
Kaina, B. (2021). On the Origin of SARS-CoV-2: Did Cell Culture Experiments Lead to Increased Virulence of the Progenitor Virus for Humans?. in vivo, 35(3), 1313-1326.
Kuo, L., Godeke, G. J., Raamsman, M. J., Masters, P. S., & Rottier, P. J. (2000). Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. Journal of virology, 74(3), 1393-1406.
Lam, T. T. Y., Jia, N., Zhang, Y. W., Shum, M. H. H., Jiang, J. F., Zhu, H. C., … & Cao, W. C. (2020). Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature, 583(7815), 282-285.
Latinne, A., Hu, B., Olival, K. J., Zhu, G., Zhang, L., Li, H., … & Daszak, P. (2020). Origin and cross-species transmission of bat coronaviruses in China. Nature Communications, 11(1), 1-15.
Lau, S. K., Li, K. S., Huang, Y., Shek, C. T., Tse, H., Wang, M., … & Yuen, K. Y. (2010). Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. Journal of virology, 84(6), 2808.
Lee, J., Hughes, T., Lee, M. H., Field, H., Rovie-Ryan, J. J., Sitam, F. T., … & Daszak, P. (2020). No evidence of coronaviruses or other potentially zoonotic viruses in Sunda pangolins (Manis javanica) entering the wildlife trade via Malaysia. Ecohealth, 17(3), 406-418.
Li, Y., Wang, H., Tang, X., Fang, S., Ma, D., Du, C., … & Zhong, G. (2020). SARS-CoV-2 and three related coronaviruses utilize multiple ACE2 orthologs and are potently blocked by an improved ACE2-Ig. Journal of virology, 94(22), e01283-20.
Li, L., Wang, J., Ma, X., Li, J., Yang, X., Shi, W., & Duan, Z. (2021a). A novel SARS-CoV-2 related virus with complex recombination isolated from bats in Yunnan province, China. bioRxiv.
Li, P., Guo, R., Liu, Y., Zhang, Y., Hu, J., Ou, X., … & Qian, Z. (2021b). The Rhinolophus affinis bat ACE2 and multiple animal orthologs are functional receptors for bat coronavirus RaTG13 and SARS-CoV-2. Science Bulletin, 66(12), 1215-1227.
Li, Z., & Zhang, J. Z. (2021). Quantitative analysis of ACE2 bindings to coronavirus spike proteins: SARS-CoV-2 vs SARS-CoV and RaTG13. Physical Chemistry Chemical Physics.
Liu, K., Pan, X., Li, L., Yu, F., Zheng, A., Du, P., … & Wang, Q. (2021). Binding and molecular basis of the bat coronavirus RaTG13 virus to ACE2 in humans and other species. Cell, 184(13), 3438-3451.
Luk, H. K., Li, X., Fung, J., Lau, S. K., & Woo, P. C. (2019). Molecular epidemiology, evolution and phylogeny of SARS coronavirus. Infection, Genetics and Evolution, 71, 21-30.
Lytras, S., Hughes, J., Martin, D., de Klerk, A., Lourens, R., Pond, S. L. K., … & Robertson, D. L. (2021). Exploring the natural origins of SARS-CoV-2 in the light of recombination. bioRxiv.
Menachery, V. D., Yount, B. L., Debbink, K., Agnihothram, S., Gralinski, L. E., Plante, J. A., … & Baric, R. S. (2015). A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence. Nature medicine, 21(12), 1508-1513.
Mou, H., Quinlan, B. D., Peng, H., Guo, Y., Peng, S., Zhang, L., … & Farzan, M. (2020). Mutations from bat ACE2 orthologs markedly enhance ACE2-Fc neutralization of SARS-CoV-2. BioRxiv.
Murakami, S., Kitamura, T., Suzuki, J., Sato, R., Aoi, T., Fujii, M., … & Horimoto, T. (2020). Detection and characterization of bat Sarbecovirus phylogenetically related to SARS-CoV-2, Japan. Emerging infectious diseases, 26(12), 3025.
Opriessnig, T., & Huang, Y. W. (2021). Third update on possible animal sources for human COVID‐19. Xenotransplantation, 28(1).
Piplani, S., Singh, P.K., Winkler, D.A. et al. In silico comparison of SARS-CoV-2 spike protein-ACE2 binding affinities across species and implications for virus origin. Sci Rep 11, 13063 (2021).
Rahalkar, M. C., & Bahulikar, R. A. (2020). Lethal pneumonia cases in Mojiang miners (2012) and the mineshaft could provide important clues to the origin of SARS-CoV-2. Frontiers in public health, 8, 638.
Shang, J., Ye, G., Shi, K., Wan, Y., Luo, C., Aihara, H., … & Li, F. (2020). Structural basis of receptor recognition by SARS-CoV-2. Nature, 581(7807), 221-224.
Sirotkin, K., & Sirotkin, D. (2020). Might SARS‐CoV‐2 have arisen via serial passage through an animal host or cell culture? A potential explanation for much of the novel coronavirus’ distinctive genome. BioEssays, 42(10), 2000091.
van Dorp, L., Richard, D., Tan, C. C., Shaw, L. P., Acman, M., & Balloux, F. (2020). No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nature communications, 11(1), 1-8.
Wang, L. F., Shi, Z., Zhang, S., Field, H., Daszak, P., & Eaton, B. T. (2006). Review of bats and SARS. Emerging infectious diseases, 12(12), 1834.
Wacharapluesadee, S., Tan, C. W., Maneeorn, P., Duengkae, P., Zhu, F., Joyjinda, Y., … & Wang, L. F. (2021). Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nature communications, 12(1), 1-9.
Wrobel, A. G., Benton, D. J., Xu, P., Roustan, C., Martin, S. R., Rosenthal, P. B., … & Gamblin, S. J. (2020). SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nature structural & molecular biology, 27(8), 763-767.
Yu, P., Hu, B., Shi, Z. L., & Cui, J. (2019). Geographical structure of bat SARS-related coronaviruses. Infection, Genetics and Evolution, 69, 224-229.
Zhan, S. H., Deverman, B. E., & Chan, Y. A. (2020). SARS-CoV-2 is well adapted for humans. What does this mean for re-emergence?. bioRxiv. doi: https://doi.org/10.1101/2020.05.01.073262
Zhou, P., Yang, X. L., Wang, X. G., Hu, B., Zhang, L., Zhang, W., … & Shi, Z. L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. nature, 579(7798), 270-273.
Zhou, H., Chen, X., Hu, T., Li, J., Song, H., Liu, Y., … & Shi, W. (2020). A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Current Biology, 30(11), 2196-2203.
Zhou, H., Ji, J., Chen, X., Bi, Y., Li, J., Hu, T., … & Shi, W. (2021). Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. BioRxiv. [Now published in Cell: Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses: Cell]
Zhou, P., & Shi, Z. L. (2021). SARS-CoV-2 spillover events. Science, 371(6525), 120-122.
If this article was useful to you please consider sharing it with your networks.

Couldn’t RaTG13 have mutated into a variant called SarsCov 2 inside a human body ?
Not likely. Initially we considered it as a possibility, but the discoveries of RmYN02 and RpYN06 and PrC31, which are in some parts of the genome closer to SARS2, suggests strongly they are closer ancestors and so more likely parents (this applies just to those parts of the genome where they are more similar, RaTG13 is the closest for the spike and some other parts). So SARS2 has many parents. I should also say that this argument is made on the basis of parsimony. It could be that what infected the miners looked very similar to SARS2 but it disintegrated (as it were) into the parts we see today in RmYN02 etc. This is equally possible, just supported by a little less evidence. Whichever way round, it doesn’t alter our basic conclusions.
This is a set of new findings based entirely on informed analysis of data in the public domain. It represents the best kind of writing by scientists in the public interest and should have a big impact on the discourse about the origins of SARS-Cov2.
By mixing these new strains in lab setting, by accident or otherwise, is it likely to generate SARS-2?
Very interesting. But seriously, why don’t you submit this to Nature/Lancet etc? This evidence needs to be presented full-on to the broader science community.
This is the best website in the world for independent scientific coronavirus related news.
Either Chinese researchers collected the brand new mosaic virus(2019 cov)from the miner’s bodies, OR somehow these closely related viruses were combined and then spread – years after the mine incident.
Jonathan – thank you and your colleagues for all the hard work in pulling this together
You concluded by saying the underlying problem is that academic science is enmeshed in a wider transnational Pandemic Virus Industrial Complex (PVIC) that has sought to suppress lab origin theories and within which the WIV and the EHA are just minor cogs.
The important consequence of this is that outbreak origin investigations are always challenging. They require people who are expert but are either not conflicted or who have demonstrated their independence.
The FOIA email information below shows this.
It sets out the concerns Chinese scienists expressed on 14th Feb 2020 that one of their senior and experienced colleagues had caught Covid-19 from lab experiments while looking into Covid-19.
These were US based scientists who were asked to write a paper showing a lab infection source of Covid-19 was not credible, but then found out how easily the Covid-19 virus could infect a senior experienced researcher in a lab environment while working in a premier virology lab in Beijing
https://usrtk.org/biohazards/senior-chinese-scientist-acquired-sars-cov-2-in-lab-infection-accident/
Senior Chinese scientist acquired SARS-CoV-2 in lab infection accident, virologist says
The SARS-CoV-2 lab-acquired infection came to light in a set of emails dated Feb 14, 2020, between virologists Shan-Lu Liu (Ohio State University), Lishan Su (then of the University of North Carolina) and Shan Lu (University of Massachusetts Medical School). The context of the email exchange was in the preparation of a commentary to refute the hypothesis that the novel coronavirus SARS-CoV-2 came from a lab, which Shan Lu had solicited as editor-in-chief of Emerging Microbes & Infections (EMI), a China-linked journal.
Shan-Lu Liu noted that his former director at NIVDC
“has now been infected with SARS-CoV-2”,
and in a separate email acknowledged that his former colleague
“was infected in the lab!”
Shan Lu responded,
“I actually am very concerned for the possibility of SARS-2 infection by lab people. It is much more contagious than SARS-1. Now every lab is interested in get[ting] a vial of virus to do drug discovery. This can potentially [be] a big issue.”
There does not appear to be any public disclosure or reporting of this lab-acquired infection of SARS-CoV-2 from the NIVDC.
This raises more questions about whether there is adequate disclosure of lab-acquired infections in China.
It also reinforces the idea that if SARS-CoV-2 originated as a lab-acquired infection at the Wuhan Institute of Virology or Wuhan Center for Disease Control and Prevention, there may not have been disclosure of such an accident.
https://usrtk.org/wp-content/uploads/2021/08/OSU-emails-LAI-SARS2.pdf
As you have set out the reluctance to provide the existing data and information
that may help lead to Covid-19s origin, even though it has been paid for by US public funding is well known.
That this involves numerous respected parties who were asked to advise the US Govt, The WHO and others (including being on formal investigation panels) about the possible origins of Covid-19 is very disturbing and set out in FOIA e-mails
https://usrtk.org/biohazards-blog/scientists-masked-involvement-in-lancet-letter-on-covid-origin/
Emails show scientists discussed masking their involvement in key journal letter on Covid origins
Daszak and two other EcoHealth-affiliated scientists thought they should not sign the statement so as to mask their involvement in it. Leaving their names off the statement would give it “some distance from us and therefore doesn’t work in a counterproductive way,” Daszak wrote.
Daszak noted that he could “send it round” to other scientists to sign.
“We’ll then put it out in a way that doesn’t link it back to our collaboration so we maximize an independent voice,” he wrote.
The two scientists Daszak wrote to about the need to make the paper appear independent of EcoHealth,
are coronavirus experts Ralph Baric and Linfa Wang.
In the emails, Baric agreed with Daszak’s suggestion not to sign The Lancet statement, writing “Otherwise it looks self-serving, and we lose impact.”
U.S. Right to Know previously reported that Daszak drafted the statement for The Lancet, and orchestrated it to “not be identifiable as coming from any one organization or person” but rather to be seen as “simply a letter from leading scientists”.
The link below has an extensive reading list of public data including the various papers written by Jonathan and his colleagues
https://usrtk.org/biohazards/origin-of-sars-cov-2-gain-of-function-readings/
Thank you
Adam
Excellent work, thank you Jonathan & Allison.
Refreshing, thorough analysis of which I hope to see more.
Regrettably, the collecting of samples may indeed have fulfilled the prophecy of pandemic. Indeed the virology/zoonotic establishment’s worst nightmare.
Are we capable of better, particularly in the face of unenlightened ‘command & control’ political systems? Let’s not only hope, but act.
As a laymanI have a number of questions for Jonathan, and I am still hoping for a responses. My questions are here: https://docs.google.com/document/d/13NE7LouunuJZnNOqiR_-e3Ip2hjNwx8lYx27UiHnrVw/edit?usp=sharing
Maybe someone else can help.
My basic question is: Isn’t it much more likely that SARS-CoV-2 was engineered (from the ingredients Jonathan described) in a lab than that those ingredients somehow (and this is the point!) hopped into a miner in the cave and somehow (again the point!) mixed themselves together into SARS-Cov-2?
The article is interesting but is so replete with qualifying speculative assumptions about “possibilities and probabilities” which in fact can not be nailed down to EVER come to a conclusion with any degree of certainty. It is fine as theory. But if we as a society remain POWERLESS to SHUT THESE SITES DOWN. It is only spinning our wheels in an intellectual web that is useless. It solves nothing, It does not help get us out of this pandemic of fear of the virus, that does not have that lethal an IFR mortality rate. Although some age groups and people with comorbidities may want to make a personal decision to take the experimental but risky vaccines given EUA by health agencies.
Hi Jonathan
Pandemic Virus Industrial Complex
We are living through difficult times and it is hard to know what is nearer the truth than is not.
There seems to have been an official approach taken about the origin of the Covid virus that involved ignoring credible information. This same attitutde also seems to apply to some treatments for Covid and their safety and effectiveness.
I know this is a minefield subject but wanted to ask your opinion as the two subjects seem related
While not seeking any medical opinion from you on the effectiveness of Ivermectin and other treatments do you see any basis for scientific concern about the approaches taken by the regulators, politicians, media, pharma businesses and medical campaigns against Ivermectin etc.
Is this all part of the Pandemic Virus Industrial Complex response ?
In support of this question here is a little of what has led me to ask about this.
There is a safety study from Merck on Ivermectin that shows it was tested at 10x the FDA approved dose and was so safe it had less adverse reactions than the placebo, yet Merck has come out saying that there is no evidence showing Ivermectin is safe at high doses.
Many links to the full report have stopped working or been removed but its on webarchive
https://web.archive.org/web/20210719231037/https://sci-hub.st/10.1177/009127002237994
Which is right what Merck says or what Merck’s own research shows?
Merck in recent comments says there is
“A concerning lack of safety data in the majority of studies. We do not believe that the data available support the safety and efficacy of ivermectin beyond the doses and populations indicated in the regulatory agency”
In 2017 Nature published a piece setting out Ivermectins many antiviral and other human mecical properties
https://www.nature.com/articles/ja201711
Below is a link setting out why some think official WHO studies into Ivermectin are being designed to fail by being given in too low doses to only high risk groups and for too short a time and not in line with successful trials
https://covid19up.org/university-of-oxford-study-ivermectin/
Here is a review of a successful Ivermectin trial
https://m.youtube.com/watch?v=BLWQtT7dHGE
It is said the opposite was done for the WHO HCQ studies which was given at toxic doses known to cause heart conditions so way above (up to 5x) recommended safety levels and for too long. Those WHO studies were stopped due to safety concerns about patient heart conditions.
Here is a review of the cancelled WHO HCQ study at high doses and an 8000 hospital patient HCQ hospital study done at WHO protocols but at normal non toxic doses showing a 30% risk adjusted reduction in Covid mortality
https://youtu.be/2uzXHnUViro
There are numerous studies showing a correlation between Ivermectin and lower Covid mortality or about Ivermectins antiviral properties
There are many more studies that indicate the same positive out comes from mass use in Mexico City, Brazil, Peru and elsewhere
https://ivmmeta.com/
I will post separately some specific links to Studies done and referred to by a range of medical groupings. I have not even linked the BIRD Group findings on Ivermectin
Studies finding no benefit from Ivermectin have often been funded by pharma companies including Merck and Gilead which are deeply conflicted. The same goes for those on FDA panels on Ivermectin
It is also worth noting the difference in FDA treatment of Remdesivir and Ivermectin seems very clear despite almost no large trials on Remdesivir or evidence of its success the FDA approved its use.
What received little coverage was a probably better and cheaper alternative to Remdedivir GS-441524 which the FDA said in 2020 looks a promising Covid treatment but slow walks FDA research and Gilead refuses to meaningfully allow independent researchers full access to the info – plus Gilead even refuses to allow it as a treatment for a 100% fatal cat coronavirus despite GS-441524 being shown 99% effective in Gilead sponsored trials and a blackmarket version has been widely used for many years
What is going on ?
Following on from the last post here are some links on Ivermectin studies
These cover both meta data of countries with long term use of Ivermectin to treat parasites studies from early 2020 and later and case studies of Ivermectin used in multiple hospitals on care workers and others showing it stopping them catching Covid.
In one trial of hospital workers in multiple hospitals over 3 months 100% remainded Covid free in IVM group while 58% became infected in the Control Group. PCR tested etc
These do not get followed up on despite involving high risk groups and having controls plus many are carried out in hospital or care home settings
A LANCET study not about IVM directly shows low Covid infection and mortality in African countries with high parasite levels. High parasite levels = high IVM use.
Ivermectin Studies Vs Covid
Ivermectin Studies showed a 10% Covid infection or mortality in African countries using Ivermectin for mass parasite campaigns compared to those that did not
African countries which distribute Ivermectin for parasites
Covid Death Rates
134.4 cases per 100,000 and 2.2 deaths per 100,000
“African countries which do not distribute Ivermectin:
Covid Death Rates
950.6 cases per 100,000 and 29.3 deaths per 100,000.”
2019, Japan’s death rate from influenza
2.9 death cases per 100,000 inhabitants.
https://www.medrxiv.org/content/10.1101/2021.03.26.21254377v1
See Chart below
https://i.redd.it/t0mkfha24bk71.jpg
2021 LANCET STUDY on African Parasites and levels of Covid
[The Lancet Study finds high levels of parasites = low Covid. This also = countries which use Ivermectin – does not mention IVM]
In conclusion, our study is the first to show a significant inverse correlation between the presence of intestinal parasites and COVID-19 severity, suggesting that parasite co-infection, with both protozoa and helminths, may protect against progression to severe COVID-19. This is corroborated by the observed low COVID-19 fatality rate in LMIC settings where parasitic infections are endemic [[11],[12]].
https://www.thelancet.com/journals/eclinm/article/PIIS2589-5370(21)00334-5/fulltext
The Tokyo Medical Association chairman compared statistics from African countries that did use Ivermectin with those that did not and Covid
https://www.lifesitenews.com/news/breaking-japanese-medical-association-chairman-tells-doctors-to-prescribe-ivermectin-for-covid/
Another USA study in early 2020 showed African countries which used Ivermectin against parasites had much lower levels of Covid than those that did not
A US based data review of countries in Africa where Ivermectin is routinely used in regular mass treatment campaigns to treat other conditions found a significant correlation between lower amounts of Covid and these mass treatment campaigns compared to countries that did not do so. Other points were highlighted in the study.
https://dryburgh.com/wp-content/uploads/2020/12/A-COVID-19-prophylaxis-Lower-incidence-associated-with-prophylactic-administration-of-ivermectin.pdf
Case Study of 3 month weekly dose of 12mg of Ivermectin tablets kept 100% of IVM group out of 1,200 Health Care Workers studied Covid free while 58% of the control group got Covid
Case study Part 1
A short 14 day study of approx 200 hospital health care workers in 1 hospital
and
Following the 14 day trial approx 1,200 hospital health care workers (HCWs) in 4 different hospitals had a 3 month trial using IVM – 12mgs of IVM tablets once a week or just used PPE.
The 200 HCWs were given Nasal Spray and Drops or just PPE for the 14 day trial
All were Covid free PCR tested
All those given IVM remained free of Covid-19 and had not previously had Covid – PCR confirmed three weeks after the trial ended
10% of the PPE group caught Covid during the 14 day trial period.
Second part of the study,
A total of 1,195 healthcare workers were recruited from four major hospitals in Argentina,
and they changed the protocol
So that the study arm used oral carrageenan and ivermectin tablets given at a dose of 12 milligrams once per week
at the end of the study,
none of the 788 volunteers treated with IVERCAR tested positive for COVID-19 during the three month study period, but
58.2% or 237 of 407 in the PPE only control group tested positive for COVID-19.
https://dryburgh.com/wp-content/uploads/2020/12/nota-journal-of-biomedical-research-safety-adn-efficacy-iota-carrageenan-and-ivermectin.pdf
French use of IVM
In early 2020 in France an elderly care home was treating all the residents and staff with Ivermectin because of a scabies outbreak
They found that during this period, seven out of 69 or 10% of the residents became ill with COVID-19, but only one resident needed oxygen. And none of the residents died. And these were high risk patients who had an average age of 90 years old.
They then looked for similar nursing homes close by that had not used ivermectin in the same timeframe.
And they found that about 23% of the 3062 residents had become sick with COVID-19 and 5% had died.
https://dryburgh.com/wp-content/uploads/2020/12/Ivermectin-benefit-from-scabies-to-COVID-19
All India Medical
This study run under WHO protocols was referred to by Japanese Drs along with a v successful trial amongst Indian Health Care Workers using low doses of Ivermectin to protect themselves before infection
A study by All India Institutes of Medical Sciences (AIIMS)-Bhubaneswar in the Indian state of Odisha found that two doses of potential drug ivermectin prophylaxis resulted in a 73% reduction in Covid-19 infection. Between 20 September and 19 October, 12 physicians of AIIMS-Bhubaneshwar conducted the study on healthcare workers (HCWs) at risk
In the two-cohort study, one set of HCWs received two-dose ivermectin prophylaxis at a dose of 300 μg / kg with a gap of 72 hours while workers in the other group received other prophylaxis.
With around 4,600 employees, over 625 employees of the institute tested positive for Covid-19.
The month-long study took place using 372 participants, including doctors, nurses, paramedics and sanitisation workers.
Based on WHO risk assessment guidelines, the contact tracing team of the institute made the list depending on subjects’ exposure to the disease.
AIIMS Director and corresponding author of the study Gitanjali Batmanabane said: “Earlier, at least 20 to 25 HCWs were getting infected with the virus daily. After the workers started taking ivermectin, the number of infection has come down to one or two per day.”
https://www.pharmaceutical-technology.com/news/aiims-study-covid-19/
https://www.medrxiv.org/content/10.1101/2020.10.29.20222661v1.full.pdf
https://www.researchsquare.com/article/rs-208785/v1
Other comments on the
Pandemic Virus Industrial Complex
Ivermectin has been used in many hospital trials and for patient use with success but is campaigned against by a range of parties
Interestingly Gilead refuses to support independent researcher trials of GS-441524 another cheaper and easier to use drug that Remdesivir converts into which then acts against Covid.
The FDA confirmed GS-441524 as promising Covid treatment in 2020 and it has been shown for many years as the only 100% effective treatment against a 99.9% fatal cat Coronavirus.
https://forums.livescience.com/threads/coronavirus-gs-441524-a-promising-covid-19-treatment-stalled-will-gilead-and-the-fda-help-texas-university-cancer-researchers-find-out-the-truth.3463/
https://forums.livescience.com/threads/please-explain-why-anivives-gc376-has-been-fast-tracked-over-gileads-gs-441524-for-covid-19-clinical-trials.3804/
https://www.livescience.com/cat-coronavirus-drugs-treat-human-COVID-19.html
Also see DoD research on Amodiaquine shown very effective against Covid in Hamsters
https://www.wired.com/story/hydroxychloroquine-is-toast-now-say-hi-to-its-dirty-cousin/
None of this is followed up on by the media
There is also the whole subject of Ivermectin poisoning which the Media fails to clarify is essentially false or ask who is starting these rumors
https://www.zerohedge.com/covid-19/rolling-stone-horse-dewormer-hit-piece-debunked-after-hospital-says-no-iivermectin
The Associated Press said 70% of poisoning calls were about Ivermectin – actually they misquoted the statement that said 2% of calls were about Ivermectin and 70% of those were about animal Ivermectin
Additionally
“85% of the callers had mild symptoms, but one individual was instructed to seek further evaluation due to the amount of ivermectin reportedly ingested.”
The alert said “no hospitalizations due to ivermectin toxicity have been directly reported to the Mississippi Poison Control Center or the Mississippi State Department of Health.”
https://www.dailywire.com/news/associated-press-adds-embarrassing-correction-to-article-claiming-70-of-calls-to-mississippi-poison-control-were-about-ivermectin-ingestion
https://www.mississippifreepress.org/15002/person-hospitalized-after-taking-livestock-ivermectin-from-feed-store-to-treat-covid-19/
https://reason.com/2021/09/06/ivermectin-overdoses-oklahoma-hospitals-rolling-stone-hoax/
So as far as can be seen in FDA FOIA documents set out in the link below there are no known proven cases of people taking FDA-approved ivermectin for COVID-19 and suffering severe and lasting ill effects. None proven.
https://covid19up.org/fda-ivermectin/
However over the counter Tylenol is killing people but the FDA not address this matter in at least the same way
In around 2017 Tylenol / acetaminophen-associated overdoses account for about 50,000 emergency room visits and 25,000 hospitalizations yearly and about 450 deaths
https://www.acsh.org/news/2017/09/11/tylenol-far-most-dangerous-drug-ever-made-11711
FDA FOIA Ivermectin
FDA information on Ivermectin Posioning – FOIA data release
FDA Pharmacovigilance retrieved 400 cases of exposure to ivermectin products.
https://covid19up.org/fda-ivermectin
Pandemic Virus Industrial Complex
From Trialsite news
Grotesque conflicts of interest on NIH ivermectin non-recommendation
NIH has also been secretive about the composition of the working group that proposed the ivermectin non-recommendation. The names of those individuals were redacted by the NIH from a document obtained through a Freedom of Information Act request for the agenda of a meeting considering ivermectin.
However, the group responsible for the ivermectin non-recommendation has been discovered through a FOIA request to the Center for Disease Control and Prevention. The FOIA response shows that the working group has nine members. Three members of the working group, Adaora Adimora, Roger Bedimo, and David V. Glidden, have disclosed a financial relationship with Merck. Merck has campaigned against the use of ivermectin in COVID-19. A fourth member, Susanna Naggie, had an extraordinary potential conflict of interest. She received a $155 million grant for the study of ivermectin following the non-recommendation. Funding for the study would have been difficult to justify if the drug was recommended for use in COVID-19. It is not known, however, if the panelist was aware of that opportunity or was planning to apply for that grant at the time of the deliberations on ivermectin.
The deception and secrecy surrounding the NIH ivermectin non-recommendation should have raised serious doubts about its integrity. The grotesque conflicts of interest of Panel members should make it clear that the NIH, as the FDA with its slandering of ivermectin as a “horse dewormer,” should not be taken seriously.
https://trialsitenews.com/grotesque-conflicts-of-interest-on-nih-ivermectin-non-recommendation/
Panel on COVID-19 Treatment Guidelines Financial Disclosure for Companies Related to COVID-19 Treatment or Diagnostics
9 panel members disclosed connections to Gilead. This same panel approved Gileads Remdesivir as a Covid-19 treatment this approval was later withdrawn.
This FDA disclosure only covers the previous 11 months of conflicts and does not disclose other non financial relationships or those that recently expired
https://web.archive.org/web/20200422210953/https://www.covid19treatmentguidelines.nih.gov/panel-financial-disclosure/
PBS has reported that
“While the worth of NIH-funded researchers’ financial conflicts of interest has not previously been made public, the U.S. Department of Health and Human Services inspector general reported last year [2019] that the value of NIH grants associated with such conflicts was $1 billion in fiscal 2018”
Its time to have full disclosure about what is is going on and how this might effect the health care medicines being offered
“This is critically important,” said Lisa Bero, a University of Sydney professor who studies the impact of corporate interests on health care research. “Many people over the years have been calling for it. We need this kind of data available.”
https://www.pbs.org/newshour/health/health-researchers-disclose-188-million-in-conflicts-of-interest-nih-data-suggests
It is worth noting 2 of the CDC / NIH Covid-19 Panels 3 co-chairs have long term links to Gilead which are hard to find and do not need to be declared or disclosed.
Some info is available in the post below
https://forums.livescience.com/threads/should-there-be-an-open-debate-and-discussions-about-conflicts-of-interest-in-federally-funded-healthcare-research.4772/
I agree with a comment above that this needs to be submitted for peer review. I would either like to see it out in the peer-reviewed literature or, if it gets rejected, see the manuscript reviews posted on this web site. If the reviews are flawed, then that would be interesting and I would like to see it resubmitted elsewhere. If the reviews are valid, then we could move on to other ideas, although I will say that this analysis seems pretty convincing, which means it would be very intriguing to see what ideas, if any, can counter it. Engaging in the process of peer review–and it is often a process requiring multiple submissions to different journals–would help to advance the science of covid origins and be of immense value to world society.
Today I was reminded again of just how critical your knowledgeable investigative. journalism continues to be. USRTK has published a COVID “timeline,” which has referenced the work you’ve done on the origin of the virus. THANK YOU. I hope you know how valuable your work is to all of us humans.
https://usrtk.org/biohazards/timeline-the-proximal-origin-of-sars-cov-2/?mc_cid=4331aeb3e0&mc_eid=7b6af43ea6
follow-up commnetnt – sorry, maybe “referenced” isn’t the right word. What the article does is expose communications between US health authorities, and shows that they were aware of your publication of proposed origin for COVID 19
Thank you Susan for sharing that new USRTK report. Best wishes Jonathan