
by Jonathan Latham, PhD and Allison Wilson, PhD
In China there is a popular joke about the southern city of Guangzhou (Canton). A visiting space alien, curious to learn about Chinese customs, tours its various provinces. Arriving in Guangzhou the alien asks the locals what their interests are. The Cantonese oblige their guest by putting the alien in a soup pot and eating it. This joke hinges on the Cantonese fondness for cooking with unusual species, many obtained from far away.
This feature of Canton’s cuisine was implicated in the original SARS (Severe Acquired Respiratory Syndrome) pandemic of 2002-04, which began in Guangzhou. It is thought that the virus arrived there with palm civets imported for speciality dishes (Wang et al., 2005).
But this culinary connection also marks a defining difference between the first SARS coronavirus pandemic and the current one. The COVID-19 (SARS-CoV-2) pandemic began in Wuhan, but Wuhan was considered a comparatively unlikely location for a natural (zoonotic) coronavirus spillover (Yu et al., 2019). It has no cultural or geographic or climatic predisposing factors.
For example, being fairly far north, bats are not abundant in Wuhan and Hubei province has few bat coronaviruses compared to hotspots like Yunnan and Guangdong (Yu et al., 2019). Unlike Canton, Wuhan is not famous for exotic fare. Nor is Wuhan near animal smuggling and trading origins (Li et al. 2019). It was for this reason that researchers from the Wuhan Institute of Virology (the WIV), which is the prime suspect in the various lab leak theories, mostly had to travel thousands of kilometres to find bats with coronaviruses (Yu et al., 2019). Furthermore, when WIV researchers needed to study a Chinese population that was not routinely exposed to bat coronaviruses (as a control group), they chose Wuhan residents (Wang et al. 2018; Li et al. 2019).
It is consequently a mystery, if SARS-CoV-2 does have a zoonotic origin, why COVID-19 should have emerged where it did. As Zheng-li Shi, head of coronavirus research at the WIV told Scientific American, in March 2020: “I had never expected this kind of thing to happen in Wuhan, in central China”.
What is the probability of a natural zoonotic coronavirus outbreak starting in Wuhan?
It is possible, and potentially helpful, to put numbers on Zheng-li Shi’s surprise. Numbers can more precisely show the incongruity of an outbreak occurring in Wuhan. But before using them it is important to specify the assumptions required so that these numbers can be treated with appropriate caution.
Such a calculation requires that we set aside momentarily all the varied, potentially important, but hard-to-quantify-and-mostly-unknown local factors, like those mentioned above, that may make certain locations or populations less or more likely to originate a pandemic. (For a broader discussion of these factors see e.g. Graham et al., 2013).
Given these provisos, and knowing that (1) bats and other animals which harbour coronaviruses are found practically all over the world, and (2) that the population of Wuhan is 11 million, and that (3) the global population is 7 billion, we can calculate the likelihood of Wuhan being the epicentre of a natural zoonotic coronavirus pandemic: The chance of a person from Wuhan being patient zero is approximately 1 in 630.
Therefore, if we were Zheng-li Shi, we would have “never expected” a natural zoonotic outbreak in Wuhan either. Imagine her surprise, and that of her colleagues when, in December 2019, they learned of a local coronavirus outbreak. They (and other researchers) travel all over the world, and not just China, looking for coronaviruses yet a pandemic breaks out in Wuhan, under their very noses. It truly is, very, very, unlikely that a natural zoonotic pandemic would start in Wuhan. Yet no commentator on the outbreak seems to have properly acknowledged the true scale of this improbability.
The second coincidence is an evolutionary coincidence
But there is, in fact, a second coincidence regarding the origin of the COVID19 pandemic. This coincidence has seemingly been entirely disregarded; but it too points strongly to a lab origin. The underlying logic is quite simple and it has to do with the evolution of coronaviruses.
Zheng-li Shi’s laboratory at the WIV is a world centre of coronavirus research. This has been mentioned often and is widely known. In particular, the Wuhan Institute of Virology is a world-leading site for bat coronavirus collection (and the virus came from a bat). But what has not been foregrounded is that, even within the coronaviruses, Zheng-li Shi’s laboratory had, of the 28 relevant coronavirus species, singled out just one of them as their special focus. And it is a member of this exact species among the 28 (called the “SARS-related coronaviruses“) that broke out in Wuhan in 2019.
This, then, is a further curious coincidence: for a pandemic coronavirus (SARS-CoV-2) to emerge in Wuhan and be a member of the species most studied at the Wuhan Institute of Virology.
The logic of coronavirus pandemics
A fuller appreciation of this coincidence requires visualising coronavirus evolution and understanding the research agenda at the WIV.
The coronaviruses are divided into four types: Alpha-, Beta-, Gamma- and Delta- coronaviruses. These are shown in Fig. 1 which is a phylogenetic (evolutionary) tree adapted from a paper by Li et al., 2020. (The print is small and so here is a link to the original figure.)
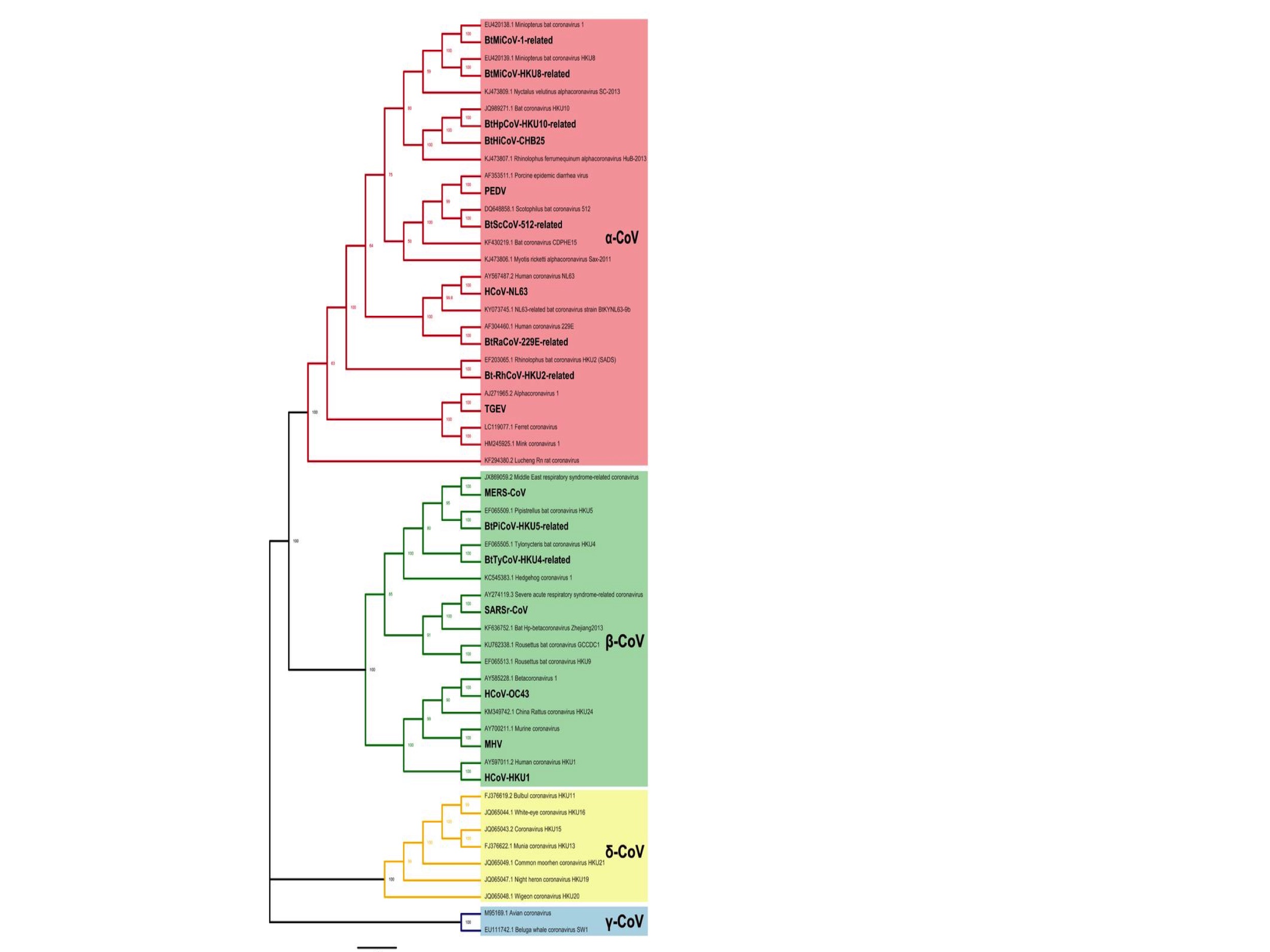
Of this phylogenetic tree, only the Alpha (pink) and Beta (green) coronaviruses will be considered here. This is because the Gamma (blue) and Delta (yellow) coronaviruses are few, not known to infect humans, and therefore questionably relevant.
As of February 2020, when Li et al. created this figure, there were 28 species of Alpha- and Betacoronaviruses. (Note: a species does not precisely equate to single tips on the phylogenetic tree in Fig 1. because some species have multiple members.)
It is important to appreciate, however, that we have no reason to suppose that a pandemic coronavirus could not have emerged from any branch of this phylogenetic tree. Indeed, the last coronavirus to jump into humans (before 2019) was MERS (Middle East Respiratory Syndrome) in 2012. MERS is a Betacoronavirus and was an unknown species before it started infecting humans. See the green arrow in Figure 2. The original SARS virus was also unknown as a species at the time it emerged as a human pathogen in 2002.
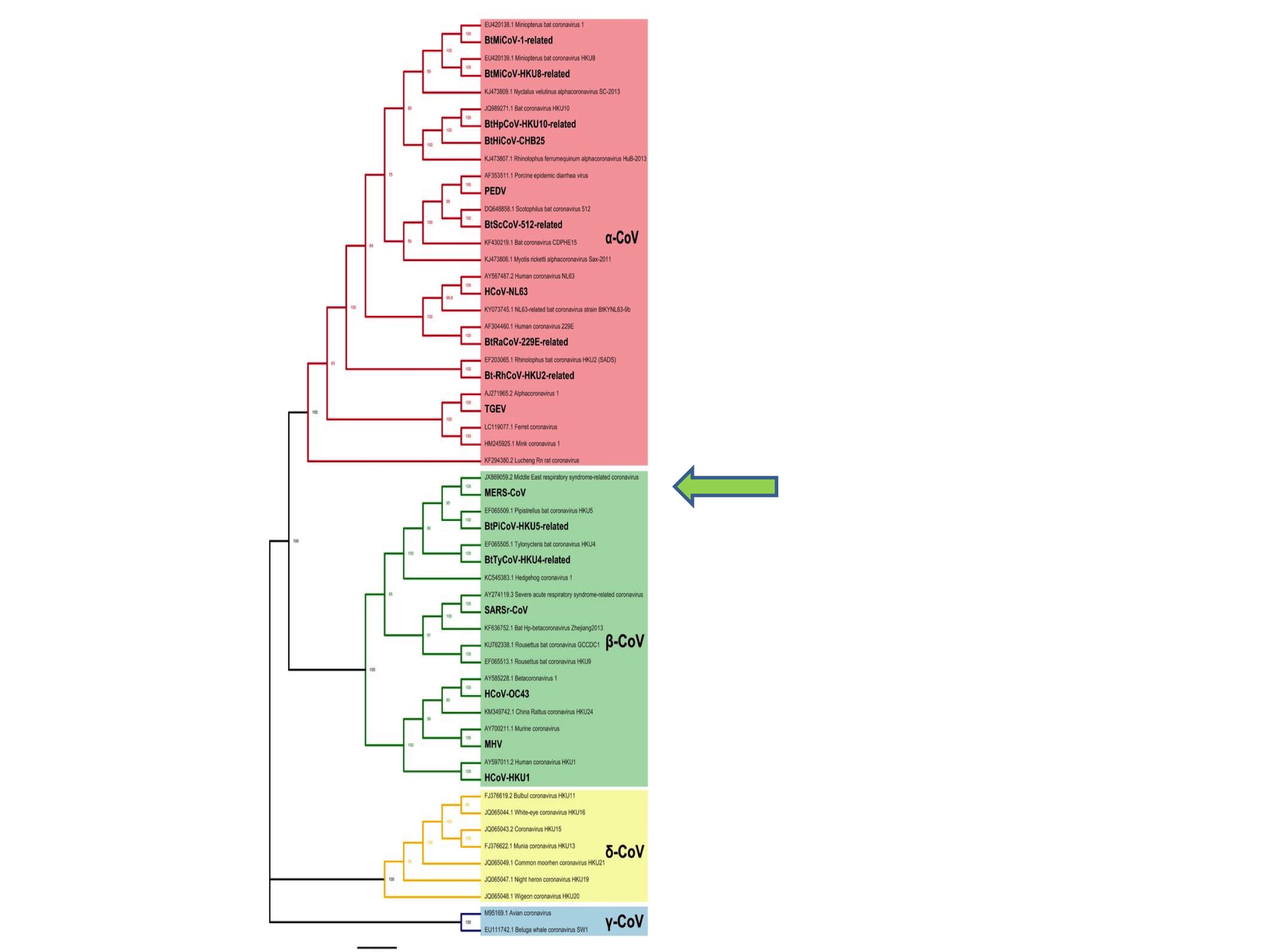
This unpredictability is also apparent from Zheng-li Shi’s choice of ‘disease X’. In 2018 the WHO announced a discussion list of pandemic priority diseases, which included Ebola, Rift Valley Fever, and other viruses. Alongside these known diseases the WHO asked experts to nominate a presently unknown candidate. Zheng-li Shi proposed that: “Disease X could be a transmissible infectious disease caused by a novel coronavirus originated from bats” (Jiang and Shi 2020). In other words, she did not predict any more narrowly than that the next pandemic would be caused by an Alpha- or Betacoronavirus.
The apparently random nature of coronavirus spillovers to humans is also apparent from inspection of Figure 3.
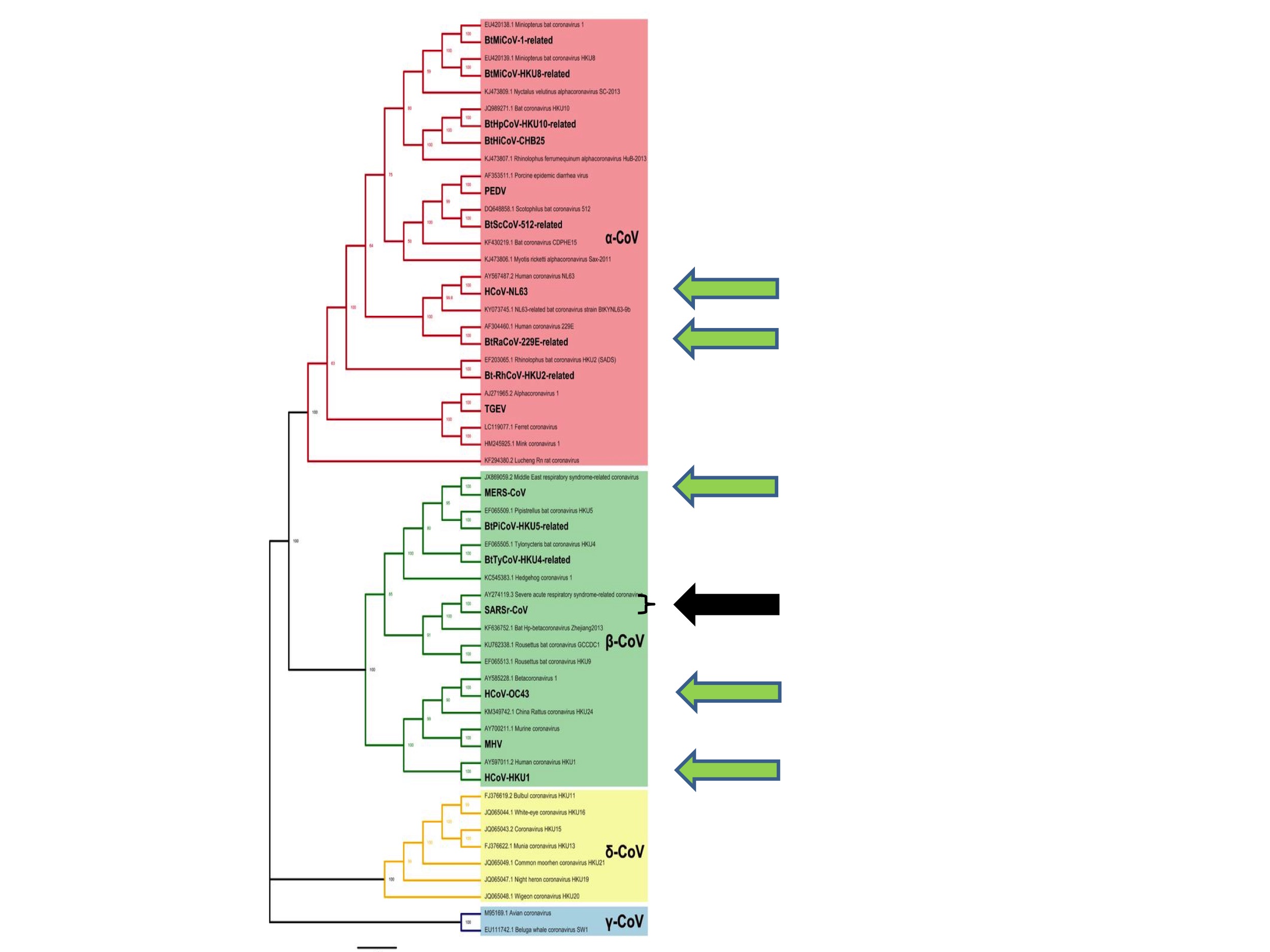
Figure 3 shows all of the six human coronaviruses identified prior to this pandemic. They are (from the top of the figure): HCoV-NL63, HCoV-229E, MERS, SARS, HCoV-OC43 and HCoV-HKU1. The six are each indicated in Figure 3 by green arrows, except for SARS, which is represented by a black arrow.
What Figure 3 illustrates is that human coronaviruses are distributed widely across the coronavirus family tree. That is to say, previous spillovers to humans happened at diverse and seemingly random points on the coronavirus tree and have involved both Alpha- and Betacoronaviruses.
The SARS-CoV-2 outbreak
With these prior assumptions stated we can then ask the question: where on the tree would one have expected (prior to the COVID-19 pandemic) the next novel coronavirus to emerge?
The answer is, if it were a natural or semi-natural spillover (i.e. a zoonosis)––from a random spot on the tree. It might have been an Alphacoronavirus or a Betacoronavirus. It might even, like MERS and SARS, be a novel species, since presumably there are still many undiscovered coronavirus species. The crucial point is that the chance of a spillover coming from each species is, as far as anyone knows, seemingly equal.
So where, phylogenetically speaking, did SARS-CoV-2 emerge?
The answer is shown in Figure 4 (below) in which the red arrow indicates the site of emergence of SARS-CoV-2.
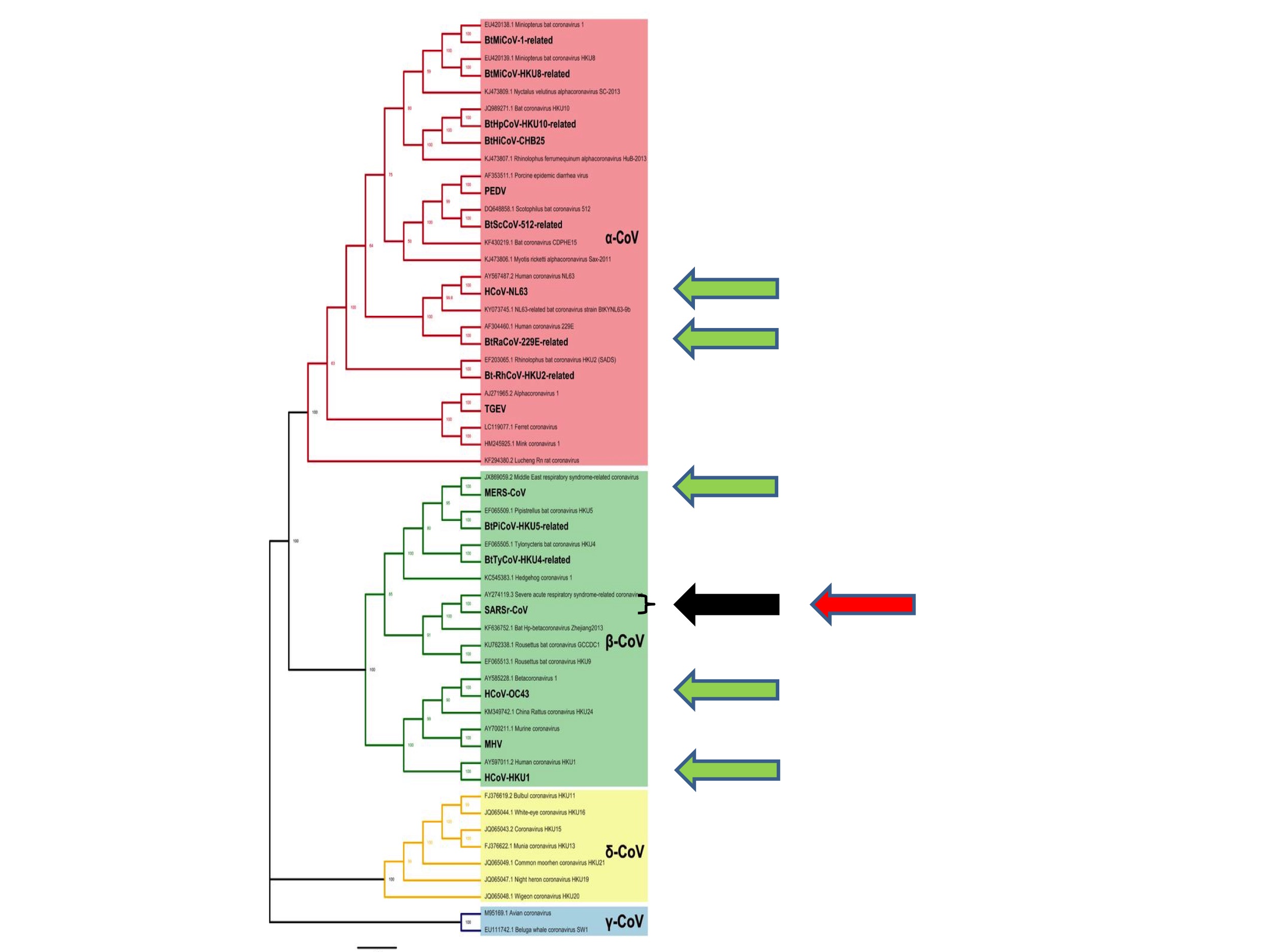
It emerged from the same species as the original SARS, hence its name. As noted above, this particular species is known to taxonomists as the “SARS-related coronaviruses” after its then most famous member (Coronavirus Study Group of the International Committee on Taxonomy of Viruses, 2020).
As discussed, from a zoonotic perspective, nothing appears to be special about these SARS-related coronaviruses. Consequently, the emergence of a second pandemic virus from the same coronavirus species constitutes a second surprising coincidence. We can again calculate its probability. If each Alpha and Betacoronavirus species is equally likely to spill over to humans, which is consistent with our understanding, then the probability of a virus from the SARS-related coronavirus species starting a zoonotic pandemic is 1 in 28. (And if there are undiscovered coronavirus species––pretty much a certainty––the number will be greater still).
It is a coincidence that, just like the emergence in Wuhan, heavily favours a lab escape if we take into account the specifics of the coronavirus research programme at the WIV, which are outlined below.
China’s research on SARS-related coronaviruses
Consider the following list of publication titles, many accepted in prestigious journals, from between 2005 and the start of the pandemic in late 2019. They are all authored by Zheng-li Shi. These eighteen research papers constitute the main focus of her published output. What they have in common is that all use the phrase “SARS-like coronavirus” or, later, “SARS-related coronavirus” or a close variant (all are bolded below). These phrases should be understood as technical terms. They denote viruses extremely closely related to SARS and only distantly related to other coronaviruses:
- ‘Bats Are Natural Reservoirs of SARS-like Coronaviruses‘ (2005);
- ‘Full-length genome sequences of two SARS-like coronaviruses in horseshoe bats and genetic variation analysis’ (2006);
- ‘Evidence of the recombinant origin of a bat severe acute respiratory syndrome (SARS)-like coronavirus and its implications on the direct ancestor of SARS coronavirus’ (2008);
- ‘Difference in Receptor Usage between Severe Acute Respiratory Syndrome (SARS) Coronavirus and SARS-Like Coronavirus of Bat Origin’ (2008);
- ‘Virus-like particles of SARS-like coronavirus formed by membrane proteins from different origins demonstrate stimulating activity in human dendritic cells’ (2008);
- ‘Immunogenicity difference between the SARS coronavirus and the bat SARS-like coronavirus spike (S) proteins’ (2009);
- ‘Intraspecies diversity of SARS-like coronaviruses in Rhinolophus sinicus and its implications for the origin of SARS coronaviruses in humans’ (2010);
- ‘Immunogenicity of the spike glycoprotein of Bat SARS-like coronavirus‘ (2010);
- ‘Bat severe acute respiratory syndrome-like coronavirus ORF3b homologues display different interferon antagonist activities’ (2012);
- ‘Identification of immunogenic determinants of the spike protein of SARS-like coronavirus‘ (2013);
- Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor (2013);
- ‘A SARS-like cluster of circulating bat coronaviruses shows potential for human emergence’ (2015);
- ‘Bat severe acute respiratory syndrome-like coronavirus WIV1 encodes an extra accessory protein, ORFX, involved in modulation of the host immune response’ (2016);
- Longitudinal surveillance of SARS-like coronaviruses in bats by quantitative real-time PCR’ (2016);
- ‘Cross-neutralization of SARS coronavirus-specific antibodies against bat SARS-like coronaviruses‘ (2017);
- ‘Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus’ (2017);
- ‘Serological evidence of bat SARS-related coronavirus infection in humans, China’ (2018);
- ‘Geographical structure of bat SARS-related coronaviruses‘ (2019).
What this list demonstrates is that, while Zheng-li Shi at the WIV focused on virus collection, above all, she dedicated her research to understanding zoonotic spillovers to humans of one species alone: the SARS-related coronaviruses.
So while most discussions of a potential lab escape have mentioned that SARS-CoV-2 emerged within commuting distance of the WIV and that researchers at the WIV worked on bat coronaviruses, none have mentioned that the coincidence is much greater than that. Zheng-li Shi concentrated, especially with her potentially highly risky molecular research, on the particular species of coronavirus that is responsible for the pandemic.
There is a simple reason for this focus. The original SARS outbreak in 2002-04 had a major impact in China. Finding the origin, explaining SARS and its symptoms, and preventing a repeat all became major research priorities for Chinese scientists.
To be sure, Zheng-li Shi published papers on other coronavirus species over that same time-period, for example on MERS, and even some on non-coronaviruses; but these articles tended to be one-offs and co-authorships with other labs. The large majority of her output and the dominant theme of her research was collecting and manipulating SARS-related coronaviruses to determine the potential for human spillover.
So, if one accepts as reasonable the assumptions made above, the probability of Wuhan being the site of a natural SARS-related coronavirus outbreak is obtained by multiplying 1 in 630 by 1 in 28. The chance of Wuhan hosting a SARS-related coronavirus outbreak is thus 17,640–1.
The criticism will doubtless be made that the geographic and the phylogenetic evidence described here are circumstantial–mere coincidences. But critiquing evidence as circumstantial is based on a common logical misconception–that circumstantial evidence represents a special category of evidence. As the philosopher David Hume first argued, all evidence of causation is composed of coincidences. All an observer can do is to add up the coincidences until they surmise that the threshold of reasonable doubt has been surpassed. Conclusions are always provisional, but in the absence of evidence to the contrary, anyone open to persuasion ought at this point to conclude that a probability of 17,640–1 far exceeds that threshold. A lab escape should at this point be the default hypothesis.
Such a conclusion is only reinforced by much of the important information that has emerged since the outbreak began. We now know, for example, that, at the time of the outbreak, Zheng-li Shi and her colleagues had in their freezers the virus sample known as RaTG13. Among all the known coronaviruses, including within the SARS-related coronaviruses, RaTG13 is by far the closest relative of SARS-CoV-2. We also know that Zheng-li Shi implied she had not actively studied RaTG13 prior to the outbreak (in Zhou et al, 2020). We now know this was false and they had been studying it since at least 2017 (Zhou et al. 2020 addendum). These facts again do not support a natural zoonotic origin.
The lack of a zoonotic theory
If there were a credible zoonotic origin theory for the emergence of SARS-CoV-2 then such a calculation might be considered moot. But, despite considerable academic discussion (e.g. Leitner and Kumar, 2020; Seyran et al. 2020; Sallard et al., 2020) and a WHO investigation, there is still no substantive zoonotic theory to speak of. Snakes, Bamboo rats, pangolins, mink, turtles, dogs, civets, whales, and frozen cod, have all, at various times, been suggested as intermediate vectors that might have carried SARS-CoV-2, or coronavirus precursors of it, to Wuhan; but neither a theory, nor a proximal spillover virus, nor a plausible intermediate host has gained significant support in the scientific community. The excellent reason is that data supporting them are largely lacking despite the apparently very intensive searching (Sallard et al., 2020).
The most concrete of these zoonotic theories, and by far the most widely known, is the pangolin (Manis javanica) theory (Anderson et al., 2020; Lam et al., 2020; Xiao et al., 2020). It is proposed that pangolins smuggled from countries to the south of China harboured precursor coronaviruses picked up from bats, thereby bringing them to Wuhan.
However, newly available evidence has made this scenario improbable. First, pangolins do not seem, after all, to naturally carry coronaviruses (Lee et al., 2020). Second, the pangolin theory rests largely on virus sequences obtained from pangolins confiscated in Guangdong province in early 2019. Attempted independent verification of these virus sequences has uncovered that, although four publications (now highly cited) discuss or report pangolin coronavirus sequences and therefore appear to support the widespread presence of coronaviruses in pangolins, only one virus genome was ever sequenced (Chan and Zhan, 2020). The papers by Xiao et al. (2020) and Liu et al. (2020) merely renamed and reconfigured sequence information generated by Liu et al. 2019. This is the same pangolin coronavirus data set discussed by Lam et al., 2020. Current thinking, in light of this new evidence, is that the smuggled pangolins were an ‘incidental host’ of the coronavirus. That is, the pangolins likely caught the virus while being smuggled (Chan and Zhan, 2020; Lee et al, 2020).
In stark contrast, there are four distinct lab origin theories and these, unsurprisingly, are getting increasing attention. Two are published in the scientific literature (Sirotkin and Sirotkin, 2020; Segreto and Deigin, 2020). A third proposes that SARS-CoV-2 was a failed attempt to develop a vaccine. This theory was developed by an independent group of online researchers called DRASTIC. The fourth is our own Mojiang Miners Passage theory.
This latter theory starts from the fact that viruses in the same mine where RaTG13 (the closest related viral sequence to SARS-CoV-2) was sampled appear to have given rise to a disease outbreak in 2012. In that outbreak, six miners were hospitalized with COVID-19-like symptoms and three died (Rahalkar and Bahulikar, 2020). All had been shovelling bat guano and were diagnosed at the time as likely suffering from an unknown coronavirus. Samples from four of the hospitalized miners were sent to the WIV for testing. To-date, there are conflicting claims about the results of those tests and nothing has been formally published (Zhou et al. 2020 addendum). The Mojiang Miners Passage theory proposes, however, that, by the time they arrived at the WIV, these patient-derived samples contained a highly adapted human virus, which subsequently escaped.
For the present moment, notwithstanding the claim of the WHO investigation and the censorship of Facebook, all of these accidental lab origin theories appear plausible to us, but all remain uninvestigated. Our prediction, however, simply based on assessing the probabilities, is that no convincing natural zoonotic origin for the pandemic will ever be found by China or the WHO or anyone else––for the simple reason that one does not exist.
References
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature medicine, 26(4), 450-452.
Chan, Y. A., & Zhan, S. H. (2020). Single source of pangolin CoVs with a near identical Spike RBD to SARS-CoV-2. BioRxiv.
S. G. of the International Committee on Taxonomy of Viruses (2020). The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nature microbiology, 5(4), 536.
Graham, R. L., Donaldson, E. F., & Baric, R. S. (2013). A decade after SARS: strategies for controlling emerging coronaviruses. Nature Reviews Microbiology, 11(12), 836-848.
Jiang, S., & Shi, Z. L. (2020). The first disease X is caused by a highly transmissible acute respiratory syndrome coronavirus. Virologica Sinica, 35(3), 263-265.
Lam, T. T. Y., Jia, N., Zhang, Y. W., Shum, M. H. H., Jiang, J. F., Zhu, H. C., … & Cao, W. C. (2020). Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature, 583(7815), 282-285.
Lee, J., Hughes, T., Lee, M. H., Field, H., Rovie-Ryan, J. J., Sitam, F. T., … & Daszak, P. (2020). No evidence of coronaviruses or other potentially zoonotic viruses in Sunda pangolins (Manis javanica) entering the wildlife trade via Malaysia. Ecohealth, 17(3), 406-418.
Leitner, T., & Kumar, S. (2020). Where did SARS-CoV-2 come from?. Molecular biology and evolution, 37(9), 2463-2464.
Li, H., Mendelsohn, E., Zong, C., Zhang, W., Hagan, E., Wang, N., … & Daszak, P. (2019). Human-animal interactions and bat coronavirus spillover potential among rural residents in Southern China. Biosafety and Health, 1(2), 84-90.
Li, B., Si, H. R., Zhu, Y., Yang, X. L., Anderson, D. E., Shi, Z. L., … & Zhou, P. (2020). Discovery of bat coronaviruses through surveillance and probe capture-based next-generation sequencing. Msphere, 5(1).
Liu, P., Chen, W., & Chen, J. P. (2019). Viral metagenomics revealed Sendai virus and coronavirus infection of Malayan pangolins (Manis javanica). Viruses, 11(11), 979.
Liu, P., Jiang, J. Z., Wan, X. F., Hua, Y., Li, L., Zhou, J., … & Chen, J. (2020). Are pangolins the intermediate host of the 2019 novel coronavirus (SARS-CoV-2)?. PLoS Pathogens, 16(5), e1008421.
Rahalkar, M. C., & Bahulikar, R. A. (2020). Lethal pneumonia cases in Mojiang miners (2012) and the mineshaft could provide important clues to the origin of SARS-CoV-2. Frontiers in public health, 8, 638.
Sallard, E., Halloy, J., Casane, D., Decroly, E., & van Helden, J. (2021). Tracing the origins of SARS-COV-2 in coronavirus phylogenies: a review. Environmental Chemistry Letters, 1-17.
Seyran, M., Pizzol, D., Adadi, P., El‐Aziz, T. M. A., Hassan, S. S., Soares, A., … & Brufsky, A. M. (2020). Questions concerning the proximal origin of SARS‐CoV‐2. Journal of Medical Virology.
Segreto, R., & Deigin, Y. (2020). The genetic structure of SARS‐CoV‐2 does not rule out a laboratory origin: SARS‐COV‐2 chimeric structure and furin cleavage site might be the result of genetic manipulation. BioEssays, 2000240.
Sirotkin, K., & Sirotkin, D. (2020). Might SARS‐CoV‐2 have arisen via serial passage through an animal host or cell culture? A potential explanation for much of the novel coronavirus’ distinctive genome. BioEssays, 42(10), 2000091.
Wang, M., Yan, M., Xu, H., Liang, W., Kan, B., Zheng, B., … & Xu, J. (2005). SARS-CoV infection in a restaurant from palm civet. Emerging infectious diseases, 11(12), 1860.
Wang, N., Li, S. Y., Yang, X. L., Huang, H. M., Zhang, Y. J., Guo, H., … & Shi, Z. L. (2018). Serological evidence of bat SARS-related coronavirus infection in humans, China. Virologica Sinica, 33(1), 104-107.
Xiao, K., Zhai, J., Feng, Y., Zhou, N., Zhang, X., Zou, J. J., … & Shen, Y. (2020). Isolation of SARS-CoV-2-related coronavirus from Malayan pangolins. Nature, 583(7815), 286-289.
Yu, P., Hu, B., Shi, Z. L., & Cui, J. (2019). Geographical structure of bat SARS-related coronaviruses. Infection, Genetics and Evolution, 69, 224-229.
Zhou, P., Yang, X. L., Wang, X. G., Hu, B., Zhang, L., Zhang, W., … & Shi, Z. L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. nature, 579(7798), 270-273.
Editor’s note. We welcome comments and information about the subject of this article. However, please note that the “reply” function in the comments section is not working for people without high level access to the website. There are two possible solutions for readers wanting to reply to specific comments:
1) Enter your comment but name the commenter you are responding to (if necessary with the date of their comment). Or,
2) Mail your comment to the editor: [email protected] and they will post it as a reply. Please be sure to say who/what you are replying too.
If this article was useful to you please consider sharing it with your networks.

Very good. But of course, US universities and military (together with EcoHealth) were also doing SARS-related CoV research, and were collaborating with WIV. Some say SARS-CoV-2 was in Europe in September 2019. Others say EVALI in the US in 2019 may have been COVID.
Hi Neil,
We do not think the other origin accounts of the virus are credible so far. A different outbreak location would be an important development, and likely disprove all Wuhan lab theories, but only if true.
Please check also the Sep 2020 paper by DRASTIC that went through a simple Bayesian argument, based on locality, and in the process highlighted all the logical and probabilistic mistakes that people so commonly make.
We also offered a simple lottery analogy, and even a prior-free argument that is very easy to understand and very convincing (generalisation of Wuhan conditions to China to appreciate the odds).
https://zenodo.org/record/4067919
When Africans demanded that the origin of HIV virus be detected we were told that it was not important and that it had nothing to do with the Belgian laboratories doing vaccine experiments on Africans in the Congo. So why the obsession with finding the source of the Covid 19 virus now? Is it because the virus is killing old white European men? Or, perhaps it has to do with Chinese economic ascendancy eclipsing the global economic dominance of the USA and Europe?
Neil, give your reference to your ridiculous assumptions then. It happened in China. The outbreak happened in Wuhan. It is ridiculous you are trying to entertain new locations of its outbreak. You obviously didn’t take this article seriously.
Politeness reminder! But yes, references are highly desirable.
Jonathan and Allison, thank you for continuing your work. I check your site often as you’ve written some of the best analysis so far of the pandemic’s origins. The fact that organizations like EcoHealth and OneHealth predicate so much of their research on spillover theory to the detriment of other potential origins of human illness …like lab leaks, industrial toxins, etc. casts further doubt on a zoonotic origin. I mention industrial toxins because there is another line of thinking that the pandemic is not as simple as one-virus one-disease; that, in effect, it may be the repackaging of multiple sources and multiple diseases. In this view, pulsed low frequency radiation from new 5G infrastructure is one culprit of many; also in this view, viruses are mistaken for exosomes, which are genetic material very similar to viruses …however which are coincident to illness, not the cause of it. Given your backgrounds, I’d love hear your thoughts on this…in other words, are you 100% certain a single virus is the cause of the disease known as Covid19? Thank you again.
Hi Mike
Every illness has multiple contributing factors. Witness the exceptional importance of co-morbidities in COVID19. And no doubt there are many other factors, especially nutritional status and toxins like PFAS. But we nevertheless believe the virus is real. Exosomes are real but they are different from viruses.
Pathogen escape from high containment laboratories do occur, not frequently, but nevertheless on a regular low incidence basis. History provides ample examples of escapes of both human and animal pathogens and it is not the intention to provide a comprehensive overview here. Such events are highly controversial because of the stakes involved by government and academia and often lead to both denial and claims of conspiracy, severely hampering a thorough investigation to confirm or reject the possibility of a laboratory leak.
The current COVID-19 pandemic is no exception to this dilemma, highlighted by the recent visit of a WHO expert group to China and Wuhan. A definitive explanation and proof of the origin of SARS-Cov-2 has not been reached until this day and may indeed never be. Still, considerable efforts are being allocated by independent scientists to study available data that might provide a clue to the origin of SARS-Cov-2 and the possible role of gain-of-function research and biosafety deficiencies.
On this day 20 years ago foot and mouth disease was first noted in pigs at a slaughterhouse in Essex, initiating what eventually became one of the biggest foot and mouth disease epidemics in history. The official inquiry into the epidemic was published in 2002 (Lessons to be Learned Inquiry Report, July 2002). This report states that ”The exact source of the FMD virus implicated in the UK outbreak will never be known.”. The chief veterinary officer concluded on the origin of the virus that ”The source of the virus for the 2001 epidemic was most probably infected or contaminated meat or meat products but it is unlikely that the origin of this material or the route by which it entered the UK and reached Burnside Farm will ever be identified”. Alternative theories of the origin of the virus were considered but, surprisingly, the possibility of a lab leak from the Pirbright Institute was not mentioned at all. Considering that the virus was almost identical to a strain isolated in South Korea in 2000, that this lineage was geographically restricted to countries in the Far East and that this virus was present at Pirbright and also being studied in animal experiments, it is surprising that the possibility of an accidental escape from the laboratory was not addressed in the official inquiry. An limited outbreak of foot and mouth disease virus in UK in 2007 was confirmed to be caused by accidental escape of virus from Pirbright. Therefore, given that it happened in 2007, it is retrospectively of concern that such a possibility was not considered in 2001.
German professor agrees, in new comprehensive study: https://swprs.org/german-study-laboratory-accident-most-likely-cause-of-coronavirus-pandemic/
From the article you linked to: “The two most recent global pandemics were the 1977 ‘Russian flu’ and the 2009 ‘swine flu’. In both of these cases, modern genetic research indicates that a lab escape was the most likely origin of the pandemic virus (see here and here). Yet in both cases, the World Health Organization (WHO) initially excluded this possibility (see here and here).”
Wow. I wasn’t aware of this.
Important article and great discussion here! For all of you who are scientists, lawyers or policy experts, please consider signing on to this
Statement by Scientists, Lawyers, and Public Policy Activists on Why We Need a Global Moratorium on the Creation of Potential Pandemic Pathogens (PPPs) Through Gain-of-Function Experiments: https://www.surveymonkey.com/r/XPJL2R9
There’s also a people’s petition here: https://advocacy.organicconsumers.org/page/18957/petition/1
Alexis Baden-Mayer, Esq, 02/19/21
I agree.
The reason this wonderful article is so important is that until we make the world aware of GOF bioweapons research, we cannot hope to gain support for regulation. We also cannot prevent likely future human created pandemics.
The same way the Atomic Age needed awareness about the problems of Atomic experimentation and warfare, the Pandemic Age needs awareness about the problems of GOF research.
If investigations on this research and lab accidents is censored, we cannot hope to keep humans safe. Thank you for providing these links.
Excellent work. The combination of these two unlikely coincidences is very convincing.
With the history of SARS in China and the Mojiang miners getting a SARS-like coronavirus disease, it would make absolute sense for the WIV to be working on a pan SARS-like coronavirus vaccine. So the Mojiang miners passage theory and DRASTIC’s vaccine gone wrong theory might come together.
The rationale of the unlikeliness that the outbreak would occur in Wuhan unless there was a lab release is kind of like the latest rationale supporting voter fraud claims: that how could Trump lose by so much if at the local level his party gained seats in the House of Representatives? Revulsion for Trump is the simplest explanation and there IS evidence for that. A lab escape may or may not have happened, but there is no evidence for it. The Military Games had 10, 000 participants from all over the world, and 250,000 local volunteers. There is evidence of the presence of the virus in Europe earlier in 2019 and in the US in December 2019. Only your final conclusion makes sense–we will probably never know.
There actually is evidence that something odd occurred at the Wuhan Institute of Virology in October 2019: https://www.nbcnews.com/politics/national-security/report-says-cellphone-data-suggests-october-shutdown-wuhan-lab-experts-n1202716
And, apparently, “The U.S. government has reason to believe that several researchers inside the WIV became sick in autumn 2019, before the first identified case of the outbreak, with symptoms consistent with both COVID-19 and common seasonal illnesses.”
That’s from: https://ge.usembassy.gov/fact-sheet-activity-at-the-wuhan-institute-of-virology/
It is possible to find out what happened if the World Health Organization can get data on the earliest cases that China is obligated to provide under the International Health Regulations. That’s what’s happening now. WHO investigators are trying to get that data and it is very important that the world back these efforts up. So far, the Biden Administration is and we should do everything we can to support that.
If COVID-19 is the result of a lab accident, it should be pretty straightforward to gather the evidence needed to figure this out. One thing the U.S. could do is a Congressional investigation where Peter Daszak and other U.S.-funded researchers involved in the work of the Wuhan Institute of Virology would be called to testify.
Would it be a stretch to say that Dr. Shi was batshit crazy?
In your article you make the statement: “to-date, there are conflicting claims about the results of those tests and nothing has been formally published (Zhou et al. 2020 addendum). The Mojiang Miners Passage theory proposes, however, that, by the time they arrived at the WIV, these patient-derived samples contained a highly adapted human virus, which subsequently escaped
The Zhou reference makes clear:
“To investigate the cause of the respiratory disease, we tested the samples using PCR methods developed in our laboratory targeting the RNA-dependent RNA polymerases (RdRp) of Ebola virus, Nipah virus and bat SARSr-CoV Rp3, and all of the samples were negative for the presence of these viruses. We also tested the serum samples for the presence of antibodies against the nucleocapsid proteins of these three viruses, and none of the samples gave a positive result. Recently, we retested the samples with our validated enzyme-linked immunosorbent assay (ELISA) against the SARS coronavirus 2 (SARS-CoV-2) nucleocapsid protein—which has greater than 90% amino acid sequence identity with bat SARSr-CoV Rp3—and confirmed that these patients were not infected by SARS-CoV-2.”
How do you reconcile your statement with the clear statement from Zhou et al that contradicts both your statement and the “passage theory”?
Two things here:
1) These statements by the Zhou et al do not actually state there was no coronavirus in the miners samples. Simply that they did certain tests that gave negative results. Let’s assume that is true but they may have done other tests that were positive. One can read the addendum as an attempt to be precise or an attempt to be evasive. Ordinarily no one would assume the latter except this addendum is precisely necessary because the authors were evasive about their research on RaTG13 in the first place.
2) Both the Master’s thesis and the PhD thesis of Canping Huang claim the WIV found coronaviruses in the miner patient samples when they were sent them. Thus two independent sources, with apparently nothing to gain from dissembling, contradict Dr Shi and the Zhou authors. That is why we wrote “conflicting claims”.
Jonathan Latham,
I read the Masters thesis. The scientific results indicate that ChengDu CDC found no SARS related virus in tested samples and a sample from one patient was identified as having IgM (no indication to what) by WIV – no mention of coronavirus. The author of the thesis was obviously enamored with SARS given that it was a hot topic at the time, as indicated by their review of the topic. Their thesis title is clear – “unknown virus” the rest is speculation based upon no presented positive scientific data. I didn’t notice a link to the PhD thesis.
ZHou makes clear that they found no Rp signals for SARS like virus, no antibodies to nucleocapsid protein and no detection of SARS CoV2. What tests would contradict these rather specific results?
Given Zhou’s data and the data from Chengdu CDC I am puzzled by your passage theory, as it seems be be based on one non specific IgM comment?
Hi Ian
The Chengdu test was for SARS itself. That is, SARS one. This test was done to exclude that possibility. It doesn’t tell if a related virus like SARS2 was/was not present. Second point, it is not “one patient”. The Chinese text does not specify the numbers. But elsewhere the text says that samples from 4 patients were taken for research, which matches the Zhou addendum statement (and the PhD thesis). Third, it is true that the test is badly described, but why would they send their sample to Zheng-li Shi at the WIV if it wasn’t to test for coronavirus? Fourth, the final conclusion that it was likely a coronavirus implies as much. Fifth, the virus came from bat guano. This also implies a coronavirus (though there are other possibilities), especially since the mine had abundant coronaviruses, including SARS-related ones.
Reply to Jonathan Latham,
The ChengDu tests were for multiple viruses, including SARS. Your statement regarding the related virus SARS-Cov2 is incorrect. You ignore the fact that in December 2019 the Wuhan outbreak was initially identified as a betacoronavirus precisely by being detected using an original SARS Test – hence there is cross reactivity.
YOU asked “why would they send their samples to Zheng-LI Shi at the WIV if not to test for coronavirus?” Bats are hosts to hundreds of different viruses, the Cheng Du tests included several different viruses – all negative. Zhou indicates that they tested for multiple viruses, including SARS like viruses and detected none.
Your fourth point is based upon a “could’ statement in a poorly written / translated Masters thesis; a hypothesis that the thesis provides no actual testing data. The IgM comment is non- specific and strange given that all patients were well past initial exposure and you would be looking for IgG.
Your fifth point is also based on assumptions rather than conclusive data. That same mine was infested with rats and evidence of henipa-like virus in the rats has been reported:
https://wwwnc.cdc.gov/eid/article/20/6/13-1022_article
In terms of bats, you are correct that there are other viral possibilities. Zhou indicates that they collected 1,322 samples only 293 contained coronavirus and only 9 of those were SARS like.
It seems that your Mojiang Miners Passage Theory ignores the ChengDu data, ignores Zhou’s report, relies on one vague sentence in a Masters thesis that is not specific – but you give it specificity based on the Masters students discussion of SARS and a conclusion of “Could be”.
Don’t you think that is a weak scientific basis to make accusations?
Dear Ian
1)Your first point “You ignore the fact that in December 2019 the Wuhan outbreak was initially identified as a betacoronavirus precisely by being detected using an original SARS Test – hence there is cross reactivity.” Yes, there is crossreactivity in some test but not others. The Chengdu test was intended to discriminate SARS from other coronaviruses. It might have cross reacted with another unknown virus but as it stands it only shows the miners did not have SARS itself. The December 2019 Wuhan tests that I know about were PCR tests (I suspect there was no antibody test then since they take time to develop) and primers were designed to pick up any coronavirus. Thus it is a non-discriminating test. Therefore the points you make are unrelated and do not contradict the argument that the miners likely had a coronavirus.
2) According to the addendum Dr Shi received samples from the miners at intervals. Given they had a persistent infection an IgM test is not strange.
3) In our original article on the Mojiang Miners thesis(https://www.independentsciencenews.org/commentaries/a-proposed-origin-for-sars-cov-2-and-the-covid-19-pandemic/) we discussed the Henipa virus findings. The authors themselves concluded that the Henipa virus they found was not the cause of the outbreak.
The main point here is that you are confusing legitimate uncertainty that the miners really had a coronavirus with the right to make “accusations”. It is not an accusation. It is a scientific hypothesis.
Lastly, yes, there are differing accounts of the tests–what tests were done and what the results were and we will be addressing this key issue. I am simply pointing out that accounts predating the epidemic are less suspect than accounts (suchas the addendum) after it began. Some of these reasons are obvious but also you are asking us to believe the Zhou authors over the others when they have already misled their readers. Hence the need for an addendum. It really should have been a retraction.
And by the way, 293 viruses out of 1,322 tests is a very high rate of coronavirus detection. It is not ‘only’.
And one last thing. It is noteworthy that, in her addendum (https://www.nature.com/articles/s41586-020-2951-z), Zheng-li Shi and colleagues did not provide any data/figures for these negative tests. It is surprising to us that Nature magazine would let her make a statement of such import (no coronavirus in the miners) without supplying the same kind of evidential support that a publication would have required. To present supporting evidence should absolutely have been a requirement.
Reply to Jonathan Latham,
I agree that the Cheng Du CDC SARS test is almost certainly a PCR test, given that this has been the standard. It is a fact that a lab in the surveillance network also using an original SARS PCR test detected SARS Cov 2 in Wuhan in December 2019.
If the Mojiang Miners were the source of SARS CoV 2, as you suggest, then there would have been a similar positive result in the Cheng Du test.
reply to Jonathan Latham,
You suggest that ZHou should have issued a retraction rather than an addendum. What in her original paper was incorrect, as to justify a retraction?
AS to providing supporting evidence in her addendum – what would you like to see?
Perhaps you should submit your theory to the rigours of a peer reviewed journal.
By the way, your comment
“And by the way, 293 viruses out of 1,322 tests is a very high rate of coronavirus detection. It is not ‘only’.”
This ignores the fact that only 9 samples were SARS like coronaviruses.
And what is your scientific basis for saying 293 / 1322 is a very high rate?
Is 9/1322 also a very high rate?
Ian Brookes: I didn’t say that the Chengdu test was a PCR test. We don’t know but I would expect it to have been an antibody test.
“This ignores the fact that only 9 samples were SARS like coronaviruses.
And what is your scientific basis for saying 293 / 1322 is a very high rate?
Is 9/1322 also a very high rate?”
Clearly most of the genetic material comprising SARS-CoV-2 derives from betacoronaviruses. However, either alpha or beta coronaviruses could have contributed to the miners infections and alphacoronavirus could even have contributed features, such as the furin cleavage site.
In the addendum the Zhou authors should have presented the data for all the negative tests (antibody and PCR) that they performed. It is standard practice in science to show photos of the blots and gels of molecular biology. Nothing strange about this request. Having the documentation trail is one of the best ways to prevent fraud.
Unfortunately at the moment peer review is being used as a gatekeeper as much as it is for quality control. Publishing in peer-reviewed journals can be extremely time-consuming, slow, and frstrating. In due course I predict that our thesis will b published there, however.
There are numerous problems with the paper. Failure to acknowledge BtCov/4991 or the miners and their disease, implying that the authors hadnt touched RaTG13 when they had. Other authors have pointed out anomalies in the underlying sequence data. See: https://www.preprints.org/manuscript/202008.0205/v3
Reply to Jonathan Latham,
It was stated that Cheng Du CDC received swabs and blood samples. Swabs are for PCR.
THE WHO International Reference and Validation Laboratory network guidelines emphasis PCR.
THe Wuhan CDC is part of the same national surveillance network and it used PCR.
No SARS detected by Cheng Du CDC is the only definitive contemporaneous test data. Supported by Zhou’s addendum.
Your theory is full of holes and misrepresentation of information. Perhaps you should be issuing a retraction.
“Swabs and blood samples can be used for either. The RaTG13 was a swab and used for PCR according to the addendum. Historically, antibody detection tests are the standard for viruses, partly because PCR is somewhat new (1990s) but also because PCR is easily thwarted by contamination.You are offering snow, Ian Brookes. Why would we misrepresent the data? What would be gained by that? We have plenty of other projects on the boil not to need to pursue ones that are “full of holes”.
Today’s CBS Sunday Morning had a 4 min clip touching on many ideas in this article:
https://www.cbsnews.com/video/bats-and-the-search-for-covids-origin/
Thanks!
Reply to Jonathan Latham
It is revealing how easily you dismiss facts.
The WHO recommended PCR tests from 2004! Is that snow?
You can’t deny the evidence and so you deflect and gaslight.
Your misrepresentation of the MSc thesis is clear. You say that the thesis says WIV found coronavirus. This is a clear misrepresentation of the statement and making speculation by the author a definitive statement.
Why do you do this? My reading of your site is that science is secondary to a political / philosophical position.
Jonathan Latham.
Referring to the Cheng Du CDC tests you say ‘swabs and blood samples can be used for either’ and then deflect with the Zhou addendum. Modifications to your original post that was referenced in my mailbox.
Provide references for a swab test being used for an antibody detection as you suggest.
Given that the Chinese CDC use PCR for SARS detection and the Wuhan test showed SARS CoV 2 cross reactivity, your theory is sunk, not just full of holes.
Hi Ian
“Referring to the Cheng Du CDC tests you say ‘swabs and blood samples can be used for either’ ”
I edited my reply to make it incorrect. Sorry, and anyway wrote in too much haste. Blood samples can be used for either and the addendum referred to tests on miner’s blood (not to anal swabs from bats). My mistake. But it was not a “deflection”.
Thank you Jonathan for such an extensive compendium of evidence on the current pandemic origin. Despite being very interesting its reading including your lively discussion, and acknowledging the comments of other commentaries as well, information we access may be polarised, obscured, and impregnated with ambition/negligence. Having reached the current state of our economies, societies, and the limitations of the public health management, it is also crucial to show ways how to advance and protect ourselves now and in the next future, either by implementing more transparent scientific practices (which is difficult for the hidden agenda of the sponsors), or by educating programmes that would contribute to autonomous management of our health. Therefore, it is in the opinion of each of us to incline to one theory over another, depending on our conditioning background we live in. Even though the reality might be very close to a lab origin (imported/exported), there will always exist factors that escape our assumptions, theories, and final conclusions. But we should not ignore in our fights the importance to find the “cure”, stay resilient, and intend to harness the effects of this pandemic as a preparatory stage for possible upcoming events. In conclusion, to you all, independent scientists, activists, and responsible individuals: it is in our interest to invest our intellect, time, energy to matters that matter and bring us closer to solutions that serve to protect our integrity.
I don’t discount the possibility of a lab escape as the source of the pandemic, but for your 1 in 28 calculation it seems to me that it would be more probable that a coronavirus spill over into humans would be closely related to a previous coronavirus. Having a close sequence relationship would also provide a shared cellular receptor. Having a previously demonstrated conduit of receptor sequence homology across animals and humans (ACE2, DPP4) I feel it wouldn’t be an even probability across all known coronavirus species but rather have some usual suspects show up more frequently. As for the 1 in 630 calculation, I guess that may be true again if all things are equal, however proximity to a previous coronavirus spill over may give a greater weight to someone living in Wuhan versus someone in Wausau. They may never find the zoonotic origin as it is a difficult task as well as your point that it may not exist. I think the jury is still out on this case.
These issues are addressed in the text. We appreciate your sharing a different points of view, however.
Blending all these ideas…
So the Mojang Miner virus turns up.
Samples are sent to WIV.
WIV discovers it is extremely phylogenetically close to SARS-1, and pursues it along the general-purpose research line of the lab – to investigate sars-like viruses that could hurt humans.
I’m a bit surprised that the ?randomly discovered Mojang virus was so phylogenetically close to SARS-1. What’s the odds of that? Is there a big natural-origin cluster of them on the tree? Did SARS-1 and the Mojang virus come from the same place?
Has a definitive source for the SARS-1 virus been found? I asked google the question. 10 years after SARS the authors of this publication didn’t seem to know. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3747529/
I should’ve looked at wikipedia that says “Around late 2017, Chinese scientists traced the virus through the intermediary of Asian palm civets to cave-dwelling horseshoe bats in Xiyang Yi Ethnic Township, Yunnan.[3]
I think Rube Goldberg would have loved the zoonotic origin theory.
Thank you for your excellent deconstruction of the official narrative.
I was surprised to find that Wkipedia’s article on batwoman Shi Zhengli contains most of the info you present despite all the misinformation banter by authorities. https://en.wikipedia.org/wiki/Shi_Zhengli
One can only assume that there is a wider agenda led by Billy Gates and One Health. One batwoman’s poison is another man’s medicine
“no convincing natural zoonotic origin for the pandemic will ever be found by China or the WHO or anyone else––for the simple reason that one does not exist.”
Correct.
“….accidental lab [release] origin theories appear plausible”
Partially correct.
You’re thinking inside the box. To get the complete answer, you need to step outside the box.
Here is a start.
The man who invented (sequenced) and then GenBanked COV2 was this man – Yong-Zhen Zhang. It was not Shi Zhengli.
Fuddman, your comments are intriguing. Could you provide more information? A search on Zhang led me to this paper, which was interesting. https://sallyparadise.files.wordpress.com/2020/12/the2ndyanreport8oct2020.pdf In summary, it proposes this was a fabricated bioweapon intentionally released, while discrediting as fraudulent RaTG13, the Mohang Miners Passage Theory, and other ideas.
Hi,
Is there any comment on the recent announcement: ‘SARS-CoV-2 detected in waste waters in Barcelona on March 12, 2019’ that is still awaiting peer review?
Thanks.
Jonathan and Allison, how does this stand up the lack of diversity in influenzaviruses? Aren’t most epidemics of seasonal influenza due to the influenza A or B viruses? What do we know about the diversity of zoonotic influenzaviruses and paramyxoviruses that have caused epidemics in the past?