
by Jonathan Latham, PhD and Allison Wilson, PhD
The task of every COVID-19 origin theory is to explain a human outbreak in Wuhan, China, when the closest wild relatives of SARS-CoV-2 are located far away, 1700 km to the South West.
In public, virologists have tended to say that the proximity of the outbreak to the Wuhan Institute of Virology, which uniquely specialises in collecting, studying, and enhancing, SARS-related coronaviruses, is a coincidence. Instead, they point to the nearby Huanan seafood market as the probable spillover site, even though it is similar to thousands of others in China.
A Huanan market origin has officially been dismissed by the authorities in China. Nevertheless, on February 25th a preprint authored by George Gao, head of China’s CDC, and 38 other Chinese virologists appeared that seems intended to settle the issue (Gao et al., 2022).
The Gao article concludes, based on several lines of evidence, including a lack of correlation between positive virus samples and stalls that sold animals, that the Huanan market was simply an amplifying event. These authors do not specify how they thought the virus did first emerge, except to note that virus samples reported from other countries predate their Huanan market sampling by several months. This conclusion is in line with Chinese government statements that SARS-CoV-2 came from outside China.
Just sixteen hours later, on February 26th, two preprints appeared simultaneously that directly contradict the Gao conclusions. The senior authors of these companion articles are an overlapping set of very high-profile virologists. None are from China.
One of these preprints asserts, based on surface swabs and other environmental samples found there, that the Huanan market was the “unambiguous epicenter” of the pandemic (Worobey et al., 2022). The second argues that SARS-CoV-2 emerged at least twice at the market (Pekar et al., 2022). According to these latter authors, one zoonotic spillover created what are known as the lineage A SARS-CoV-2 viruses and a second spillover was the root of all lineage B SARS-CoV-2 viruses. These two spillovers, they say, decisively contradict a lab leak.
Many SARS-CoV-2 genomes found early in the outbreak are intermediate in sequence between the A and B lineages. These intermediates have previously been assumed to indicate a single spillover with one lineage evolving into the other (Morel et al., 2021; Pipes et al., 2021). Pekar et al. propose instead that such intermediates are all either artifactual (mostly sequencing errors) or are otherwise irrelevant to the origin. Both papers are coy, however, about what type of animal was involved in their theorised spillovers.
These conflicting conclusions set up an interesting dynamic. Clearly, Chinese virologists do not support the market hypothesis. On the other hand, the senior authors of Pekar et al. and Worobey et al. are prominent Western virologists. Many, like Kristian Andersen, Robert Garry, Ed Holmes, and Andrew Rambaut, are vocal public supporters of a zoonotic origin, and very close to Anthony Fauci the director of NIAID.
One aspect of this dynamic is the East/West split. Clearly, the two factions are not co-operating. The other is a difference of approach. The Chinese researchers assert what they think did not happen. In contrast, by formulating an explicit hypothesis (except for the host animal), the Western virologists have staked their credibility on a specific theory. The former approach is low risk; the latter is high risk since any specific theory is potentially vulnerable if new evidence were to come forward that disproves it (as proven earlier cases would); but the reward has been ample media attention of the “The Lab Leak is Dead” type.
A significant feature of this episode is the close timing of the three papers. If more evidence was needed of non-cooperation, it seems obvious that the Worobey/Pekar preprints were an ambush. Their appearance was precisely timed to spike the headlines that the China CDC paper would likely have generated by ruling out a market origin.
Do Pekar and Worobey make their case?
Especially if one includes the new Gao et al. evidence, there are already powerful reasons to doubt both the market-zoonotic origin and a dual-spillover. These reasons are largely glossed over by the Pekar and Worobey preprints so they are worth outlining briefly:
1) The market samples were probably taken late in the Wuhan outbreak.
The first reason, and the simplest, is that the market samples were taken between Jan 1st and March 30th, 2020. Yet plentiful evidence, such as contemporaneous newspaper reports of an outbreak in Wuhan, implies that SARS-CoV-2 was circulating widely in Wuhan and beyond by January 1st. Such evidence makes it difficult to agree that the market samples, so hotly discussed, have any special relevance to the source of the pandemic virus itself.
For example, according to the WHO COVID origin investigation, there were 174 COVID-19 hospitalisations in Wuhan by December 31st 2019. Given the normal delay between infection and hospitalisation and the significant rate at which COVID-19 gives asymptomatic and mild cases, these hospitalisations likely represented only the tip of a large infectious outbreak in December.
Indeed, Ian Lipkin, an epidemiologist at Columbia University, told an interviewer he knew of an outbreak in Wuhan by December 15th, 2019. Lipkin has subsequently confirmed this statement. And in spring 2020, Peter Daszak, President of the EcoHealth Alliance, Marjorie Pollack, an epidemiologist who runs ProMED, and Public Health Professor Lawrence Gostin made similar statements to the LA Times. Further back still, according to ABC News, US security agencies were tracking a pneumonia outbreak in Wuhan in November.
Early wide spread of the virus in Wuhan is evidenced too by a detailed case study of a family from Guangdong who visited Wuhan between December 29th and January 4th, 2020. Five from a total of six family members contracted COVID-19 while in Wuhan, without them having visited any markets (Chan et al., 2020). Further afield, a significant body of genome sequence and antibody evidence suggests SARS-CoV-2 was in Europe and other countries in the fall of 2019, well before the Huanan market samples were taken (reviewed in Canuti et al., 2022).
If there were thousands of cases in the city of Wuhan by Jan 1st when the market was closed and 10,000 people per day usually visited it, how do samples taken then (or later) constitute credible evidence for a market origin? Quite likely, vendors and others at the market found to have COVID-19 infections were just typical for Wuhan in December 2019 (Courtier-Orgogoz and de Assis, 2022). Typical or not, the market samples were collected too late to distinguish a market origin from any other origin in or near Wuhan.
2) Environmental samples collected at the market are of human origin and did not come from animals sold there.
The aim of the Chinese CDC paper was to analyse the environmental samples (swabs from surfaces etc.) that they took in and around the Huanan market after Jan 1st, 2020 (Gao et al., 2022). They concluded that the market was only an amplifying event, in part because SARS-CoV-2-positive samples were associated with stalls belonging to multiple types of vendors, including those not selling animals (the Worobey preprint argues there is a correlation). More compelling, the CDC authors found that the samples collected from the market, which Pekar and Worobey claim are from infected animals, are admixed only with human genetic material and not with genetic material from raccoon dogs or other species potentially sold at the market. The only reasonable inference is that these positive samples did not derive from the faeces or urine or exhalations of a live non-human animal. Few results would better indicate that virus-positive market samples derive from infected humans as opposed to other species.
3) Pekar and Worobey rely on circular reasoning to identify root viruses.
The 2022 Pekar et al. preprint adapts the findings of a previous publication (Pekar et al., 2021) to generate the novel hypothesis of a split phylogeny that traces SARS-CoV-2 back to two independent spillovers, both occurring in the Huanan market. These two spillovers, they claim, are represented today by what are known as lineage A and lineage B viruses, which differ by only two mutations. However, the phylogenetic methods used for building evolutionary trees and thus identifying the root virus in both Pekar papers are highly problematic because they are vulnerable to uneven and biased sampling and unusual genetic phenomena, such as superspreading events (Liu et al., 2020). One key bias relevant here is that, for many early COVID-19 cases, contact with the Huanan market was a diagnostic requirement (Liu et al., 2020). This will tend to orient phylogenies towards the market. Further, Pekar et al. use a clock-based algorithm that uses sampling dates to infer the root virus. This method is designed to channel the choice of root virus towards those genomes sampled earliest. If the market was the early focus of sampling, which it was, then the Pekar method of inferring the root is based on two independent forms of circular reasoning. These biases were further amplified by the Pekar and Worobey authors, who themselves decided, based on scant evidence, which patient cases counted towards the dataset and sometimes what were their disease onset dates. The effect of this intervention was to add yet more circularity into the selection process for root viruses. To satisfactorily determine which viruses are closest to the true origin requires instead a different method, one that is explicitly independent of ascertainment biases and subjective decision-making (Liu et al., 2020).
4) Pekar et al., lack the evidence for two spillover events.
A key assertion of the Pekar preprint is its proposal that extant lineage A and lineage B viruses represent the descendants of two independent SARS-CoV-2 spillover events (Pekar et al., 2022). To succeed, this double spillover claim must explain why numerous genome sequences exist that are intermediate between lineage A and lineage B. To overcome this challenge, Pekar et al. propose that such intermediates are all either artifacts from sequencing errors or irrelevant to the origin question for other reasons. Sequencing errors are common enough, but Pekar et al. only demonstrate them convincingly in a minority of instances. For example, for most of their suggested sequence artifacts they rely on an unverifiable ‘personal communication’ from a single scientist (L. Chen) in China. To make a case for the irrelevance of others they have to suggest, for example, that two genomes sampled in February in Beijing are irrelevant–as if early sequences cannot have spread elsewhere or persisted. Ultimately, their bold suggestion that the phylogeny of SARS-CoV-2 is best explained by resolving it into two independent spillovers is very poorly supported by evidence.
The Great Virological Game
Pekar and Worobey fail to make their case and hence it is tempting to dismiss their heavy reliance on weak data, their non-parsimonious interpretations, their cherry-picking, and their circular reasoning as simple shoddy science. But, in our experience, this would be an error. Poor science by otherwise competent scientists, and on such a scale, usually happens for a reason. And, in the light of the meeting, described by Katherine Eban of Vanity Fair, between Jesse Bloom, Kristian Andersen, Anthony Fauci, Francis Collins, and others, which revolved around deleted sequences from early patient samples, it seems more clear than ever what that reason is.
Ordinary bad science mostly occurs for reasons that are simple and mundane. Perhaps a research field is considered a scientific backwater to which second-rate researchers have gravitated, or a research thesis was badly supervised or not completed. If so, the results are almost sure to appear in a low-ranking peer-reviewed journal.
The other class of bad science fits a very different pattern. It sometimes occurs that the leaders who control science’s purse-strings commit to a theory or a major programme that is then contradicted by emerging evidence. If, for political or financial reasons, a policy course correction is unavailable, a rationalisation in a prominent scientific journal will be necessary to provide, as the Vatican might put it, “guidance for the faithful”.
Such publications typically have an unnecessarily large number of authors, who will mostly be laboratory heads and other prominent scientific leaders; the article will usually appear in a journal with the very highest visibility, like Nature, Cell, or Science, and the mistakes such papers contain (the rationalisations) are never errors–they are purposeful and carefully calculated. A classic of this genre, which we dissected in detail, was the NIH response to the failure of the human genome project to deliver on its promise of explaining human noncommunicable disease, a problem that remains to this day (Manolio et al., 2009).
Obviously, lab leak theories are a top concern of the infectious disease community and coalescing opinion around a semi-plausible zoonotic hypothesis is the rather obvious intent of Fauci’s NIAID. Granted, we don’t yet know in which journals Pekar et al., 2022 and Worobey et al., 2022 will appear, but their precursors (Pekar et al., 2021 and Worobey, 2021) were both published in Science. Before that, the prototype for all future efforts was The proximal origin of SARS-CoV-2, published in Nature Medicine (Andersen et al., 2020).
Putting this all together, one can see that both Chinese and Western virologists are pursuing the same general strategy, the first step of which is to ignore, discredit, deny, delete, destroy, or otherwise conceal, early sequences and samples (Bloom, 2021; Canuti et al., 2022). Erasing, or failing to collect, early information has the primary effect of lessening the likelihood that a true origin will ever be retrieved. Secondarily, erasure also makes it easier to force preferred conclusions on the data that remain. On the Chinese side, removing or failing to collect early evidence of a Wuhan outbreak helps to place the first documented appearance of the virus outside of China entirely. The Western goal has instead been to force a market spillover conclusion by preferentially discrediting samples and cases that occurred prior to the market sampling and those with no links to the market (Pekar et al., 2021 and Worobey, 2021).
These strategic aims would be unfeasible without the circularity of the standard phylogenetic methods discussed above, which are quite widely understood by insiders (Liu et al., 2020; Kumar et al., 2021). Thus, picking and choosing early samples lets clock-based phylogenetic methods that are vulnerable to sampling biases deliver a foreordained root virus.
The major complication, visible to all after the publication of the conflicting Gao, Pekar, and Worobey preprints, is that a significant conflict has arisen due to the divergent origin scenarios each group is trying to fit the facts to.
It is a reasonable inference from the above that the leading virologists on each side, and who are directing these efforts, strongly suspect (or know because they are sitting on the evidence) that early data would not exonerate virus research in Wuhan, otherwise the same people would be hunting for early samples with great alacrity, which is clearly not happening. And we can surmise it is a lab leak that is being covered up since it is the only concern that both Chinese and Western virologists could plausibly share.
Until recently, the COVID-19 origin question was therefore set to devolve into a simple two-way tug-of-war between the Gao club in the East and the Fauci club in the West. What none of them expected, however, was that a novel phylogenetic method would emerge capable of discrediting their careful calculus.
Mutational Order Analysis
Recently, a different method has been applied to the SARS-CoV-2 origin question (Kumar et al., 2021). This method is new to virology but it is widely used in cancer research (e.g. Miura et al., 2018). Using it, Kumar, Pond, and colleagues were able to infer the existence of viral strains that are older (i.e. ancestors of) Wuhan-hu-1 (the standard SARS-CoV-2 reference genome) and the other market sequences by at least 3 mutations, which is a lot.
Their innovative method is called Mutational Order Analysis (MOA). MOA is an important advance over standard approaches, not least because it doesn’t rely on clocks (i.e. time) to orient (i.e. bias) the evolutionary trees it produces. Rather, it uses genome sequence data alone to deduce the progenitor virus. Thus, MOA can be used to undo known biases, such as clocks, other sampling confounders, or even systematic sample destruction.
To understand the major difference between MOA and the phylogenetic method used by Pekar et al., consider a theoretical individual in Wuhan in late 2019 who caught a very early case of SARS-CoV-2 and who then flew to a distant country. Once there, they seeded a minor outbreak that lasted just a few weeks or months (not unlike the Guangdong family noted earlier). If any genome from a later case in this outbreak were by chance sequenced, this information would be very valuable. It would be a rare example of a root virus genome (or very close to it). From this hypothetical example we can see that even sequences that occur late in an epidemic, or far from a presumed geographic origin, can, in principle, preserve critical information about that origin.
Ordinarily, phylogenetic analysis of the origin (including Pekar et al., 2021 and 2022 and Bloom, 2021) tends to focus, sometimes entirely, on early sequences and those found local to the outbreak origin. Genomes considered of improbable relevance to the origin question are ignored. For instance, Pekar et al. 2022 performed their analysis on 787 genomes, with a cut-off date of February 14th, 2020. MOA, however, can use every available genome sequence to build up a picture of viral relatedness, without discriminating. If any virus genome in the data set constitutes an apparent missing link between two viruses it will be inserted as part of the mutational order that is built up. Because MOA uses a very large data set, specifically to capture idiosyncratic events such as the one theorised above, it can build a far more accurate, far more detailed, and much more statistically robust evolutionary tree than conventional approaches. Best of all, it does so without introducing any biases of its own.
For these reasons, MOA is clearly a superior method. Its value is especially great for inferring outbreak origins in cases where, like SARS-CoV-2, early sequences are scarce and their collection is subject to sampling biases.
MOA contradicts the Pekar and Worobey dual spillover market theory
1) MOA identifies just one root virus.
From almost 176,000 full-length genomes, MOA was able to decipher a mutational order for the origin viruses, with very high statistical confidence (Kumar et al., 2021). Their results are summarised in Fig. 1 below (taken from Fig. 2 of Kumar et al., 2021).
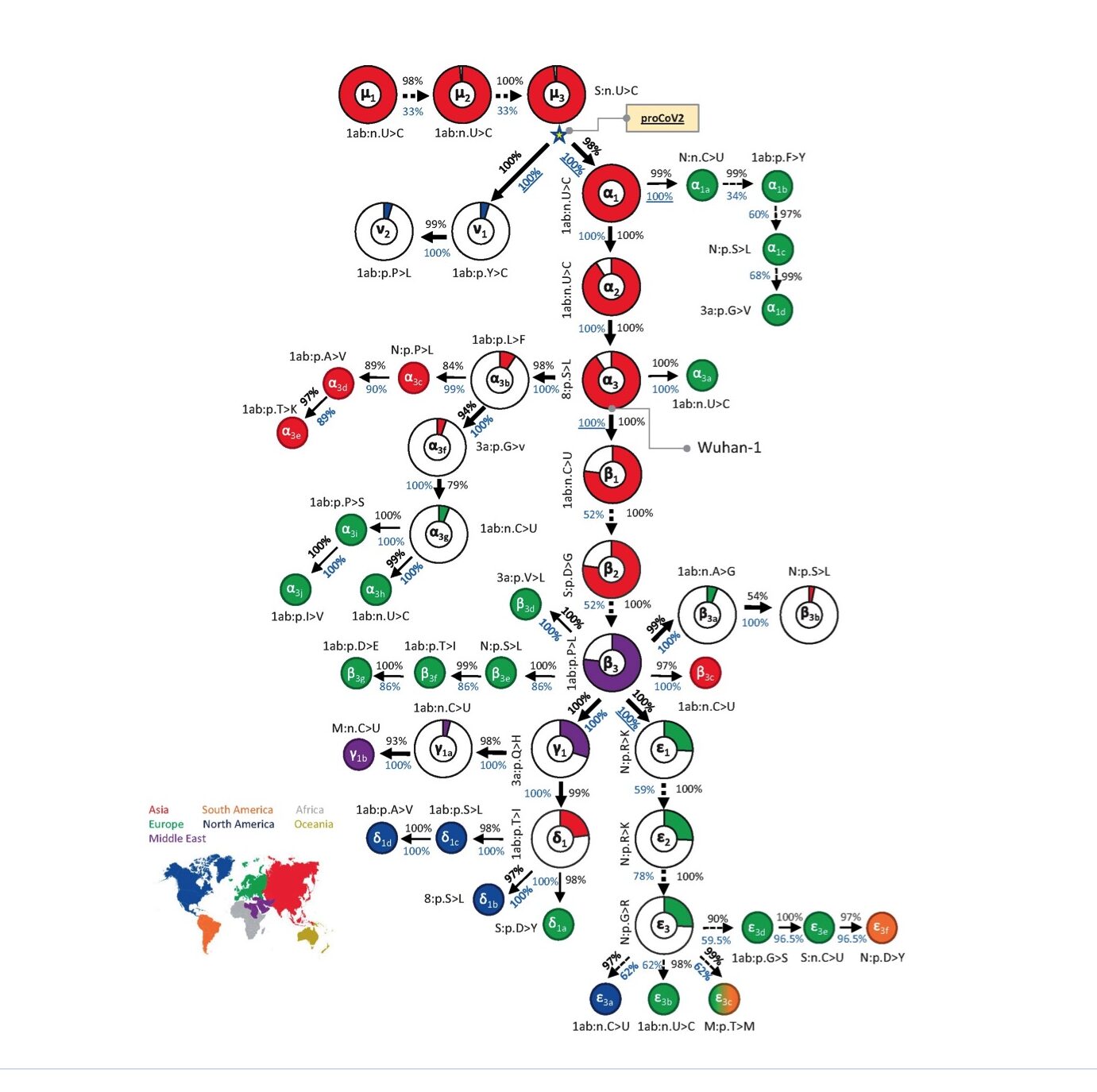
Overall, three of its findings strongly contradict the dual spillover market theory:
1) As is apparent from Fig. 1, unlike Pekar et al. (2022), MOA identifies a single root virus (μ1, top left, is its first mutant). A single root virus means the pandemic began with only one initial spillover. A single spillover event is a crucial observation because it strongly implies a lab leak (since scientists tend to work with pure cultures); whereas equivalent evidence for multiple and/or genetically diverse spillovers would have implied a natural source. MOA also shows that all lineage B viruses are descended from one lineage A virus.
2) The root virus identified by MOA predates all the root viruses identified by Pekar and Worobey.
The virus identified by MOA as the root is separated by multiple mutations from any of the viral genomes found at the Huanan market and from Wuhan-hu-1. This infers that all known market samples are well downstream of patient zero. Therefore too, as was also concluded by Gao et al., the market samples represent an amplifying event, at most.
Evolution takes time to occur. Because multiple mutations accumulated before the first confirmed cases, Kumar et al. calculated that the initial spillover (presumably in Wuhan) was around late October/early November, i.e. several months before the Wuhan market was first sampled (on Jan 1st, 2020). This conclusion is hence consistent with the presence of far-flung SARS-CoV-2 genomes in Europe and elsewhere in the fall of 2019. It is also consistent with wide spread of the virus in Wuhan before the market samples were collected.
Very recently, Kumar and colleagues released a further preprint (Caraballo-Ortiz et al., 2022). It uses even more data (1 million genomes) and an improved method, which they call TopHap, to make their findings still more robust. The addition of many more genome sequences, including some very close to the root, affirms the original conclusions of a single root virus and that lineage B evolved from lineage A. It also allowed them to move back the root virus by one further mutation and this pushes back even further in time the predicted date of SARS-CoV-2 emergence–into September, 2019.
This September spillover date further contradicts Pekar and Worobey’s date. However, it agrees well with a broad set of other phylogenetic analyses and makes the farflung virus findings in Europe and elsewhere more plausible still (Mostefai et al., 2021; Schrago and Barzilai, 2021; Song et al., 2021; Xia, 2021).
3) Analysing only viruses found in China determined the results of the Pekar articles.
In their phylogenetic analyses, both Pekar papers are noteworthy for selecting narrow geographical and temporal cut-offs for their data sets. Though these choices seem, at first sight, reasonable enough, as Kumar et al. point out for Pekar et al., 2021, the choices determine their ultimate conclusions. The excluding of sequences obtained outside China (and also those sampled more than 4 months after December) prevented these authors from including the major early branch that began with v1 (see Fig. 1). The v lineage was first sampled in the US but is needed for the correct selection of a root virus (see also Morel et al., 2021). Ultimately, the complete exclusion of the v lineage due to use of narrow cut-offs is a key reason why the two approaches reached divergent conclusions (Kumar et al., 2021). In short, the scientific contribution of Pekar et al. (2021 and 2022) is to deploy narrow windows for data acquisition to accomplish the phylogenetic equivalent of p-hacking.
If Pekar and Worobey are wrong, are Gao et al. right?
The MOA method is only somewhat kinder to the Gao et al. origin conclusions.
Although MOA supports the market being a secondary site, it contradicts the idea of a virus origin outside China. Gao at al. point at evidence for very early cases outside of China and imply that one of these was the ultimate source. But, it is clear from the Kumar et al. analysis that Wuhan and China are where the genetic diversity around the root occurs. In other words, the virus is unlikely to have emerged in Italy (or elsewhere) in late 2019 and seeded Wuhan. Far more probably, it spilled over in Wuhan and seeded Italy.
The status of a zoonotic origin in the light of MOA
We described above seven major flaws in the hypothesis of a double market spillover. Most of these criticisms apply to any hypothesised spillover scenario at the Huanan market, but it is notable that the MOA findings of Kumar, Pond, and colleagues advance the critique very significantly. Their two papers thus represent important landmarks in the study of the origin of COVID-19 (Kumar et al., 2021; Caraballo-Ortiz et al., 2022).
The key points are worth recalling: The extensive evidence of an epidemic in Wuhan long before the Huanan market samples were taken; that the market samples containing SARS-CoV-2 were mixed with human RNA and not with animal RNA; the circular reasoning of Pekar et al.’s phylogenetic method; the lack of support for dismissing viruses intermediate between lineages A and B; the methodological superiority of the MOA method, which identifies a single and significantly earlier root; and, last, the dependence of the Pekar evolutionary tree on ignoring SARS-CoV-2 sequences harvested either late or outside of China.
There is also the wider context to consider. In case it needs reiterating, there is still no evidence for wild or farmed animals being infected with SARS-CoV-2 in China, either before, during, or since the pandemic broke out. Moreover, zoonotic theorists have been very reluctant to specify a clear candidate animal species as an intermediate host. This seems to be because it is hard to construct a good case for any of them. Third, there is the unremarkable nature, by Chinese standards, of the Huanan market. Why Wuhan? It is a question still unanswered by natural zoonotic theories.
In short, it is unreasonable to claim that the Huanan market was the “unambiguous epicentre” of the pandemic (Worobey et al., 2022). Such certitude is scientifically unwarranted and only serves to give the appearance of a false flag operation.
This impression is strengthened since neither Gao, Pekar, nor Worobey, ever discuss the existence of the Wuhan Institute of Virology (WIV) and the obvious alternative hypothesis that they know goes along with it. The WIV is just a few miles from the market and, according to its US funders and the people who work there, it specialises in the collection and study and enhancement of SARS-related coronaviruses (Latinne et al., 2020). For decades, a major goal of its research has been to identify or create ones primed for human spillover (e.g. Li et al., 2019).
MOA, a goldmine of pandemic origin information
The two MOA/TopHap papers provide by far the strongest candidate yet for a root virus and they detail its subsequent evolution (Caraballo-Ortiz et al., 2022; Kumar et al., 2021). Consequently, they have even more to tell us because precise rooting is invaluable for understanding additional key aspects of any virus origin.
One key question to ask any zoonotic origin hypothesis is whether the root virus could infect putative intermediate hosts, such as the raccoon dog suggested by Pekar and Worobey. Answering this requires knowing precisely what those early strains were. Such tests can be highly misleading if performed on virus strains from much later in the pandemic since the variants of SARS-CoV-2 have distinct mammalian host ranges (Montaguteli et al., 2021; Gu et al., 2020). In particular, it ought to trouble origin theorists that the only evidence for SARS-CoV-2 infecting raccoon dogs comes from a later (D614G) strain (Freuling et al., 2021).
A second benefit of accurate rooting is probably the most significant of all. It follows from the fact that root viruses can show what, if any, were the initial adaptation steps of SARS-CoV-2 to humans.
For example, the MOA phylogeny indicates (though this can’t be seen in Fig. 1) that even the very early virus strains that predated the market samples, persisted unchanged long into the pandemic. This is a very important observation. It shows that, notwithstanding later improvements in its fitness, SARS-CoV-2 was very well adapted to humans by September or October and, so far as we can tell, from the very start.
Corroborating this, the very earliest strains differ only by synonymous mutations (see e.g. mutants μ1-3 in Fig. 1). Synonymous (as opposed to nonsynonymous) mutations are nucleotide changes only; that is, they do not alter the amino acid sequence and so they usually have zero effect on the fitness of the virus.
(Note: For each mutant, Fig.1 shows either nucleotide changes (>) or amino acid changes (>). Nucleotides are represented by the letters A, C, G, or U and changes to these represent synonymous mutations; other letters represent the standard amino acid notation system and these letters thus indicate nonsynonymous mutations.)
From the synonymous nature of the earliest mutations, the authors concluded that these earliest viruses:
“already possessed the repertoire of protein sequences needed to infect, spread, and persist in the global human population” (Kumar et al., 2021).
Moreover, many immediate descendants of these viruses often also contained only synonymous mutations and yet these strains too often became abundant. Indeed, such barely altered genomes were found “on every sampled continent” (Kumar et al., 2021).
Again, this implies the same conclusion. Whichever way one looks at the evidence, it is evident that even the very earliest viruses were not only highly adapted to humans but able, unaltered, to cause a pandemic.
This is a tremendously informative result. The question of whether SARS-CoV-2 was highly adapted to humans (and thus presumably preadapted) has been in hot dispute almost from the beginning of the pandemic (Zhan et al., 2020). But the MOA phylogeny provides by far the most decisive evidence yet that indeed it was.
Reading into their phylogeny a little further gives even more support for preadaptation. It was not until the eighth mutation did one arise (β2 in Fig. 1) that has subsequently been shown to increase the fitness of the virus in humans (Dearlove et al., 2020; van Dorp et al., 2020). β2 is the well-known D614G mutation, first identified in Wuhan late January. By this time the pandemic was well underway.
The only other mutation in Fig.1 that became abundant during the pandemic was its immediate predecessor, β1. β1 is a synonymous mutation (again) that probably hitch-hiked on D614G (Dearlove et al., 2020).
The significance of this pattern is to indicate that, even though they gave amino acid changes, even the nonsynonumous mutations clustered at the root were selectively neutral. That is, they too arose at random and not because they conferred any advantage on the virus. This again strongly reinforces the idea that the virus was under little pressure to adapt during its early spread in Wuhan.
It is not at all normal for a virus to enter a new host population without also evolving rapidly to adapt to it. Thus the first SARS coronavirus (SARS-CoV) acquired amino acid changes during its early spread in humans (Zhan et al., 2020). The alternative norm is for the virus to fail to adapt to its new host at all, such as has happened so far with every one of the many introductions into humans of the coronavirus MERS (Dudas et al., 2018).
Conceding that SARS-CoV-2 was preadapted, some have argued that SARS-CoV-2 is a ‘generalist’ virus (Frutos et al., 2020). There seems to be very little evidence for this. SARS-CoV-2 does not infect most mammal species (Kock and Caceres-Escobar, 2022). It can be actively transmitted by far fewer species still, and, when those few do transmit the virus, adaptive mutations occur (Gu et al., 2021; Sawatzki et al., 2021; Tan et al., 2022). In short, SARS-CoV-2 has not proven preadapted to any mammalian host species tested so far (except humans) and so it bears none of the hallmarks of a generalist virus.
Preadaptation of SARS-CoV-2 implies a lab leak. But it also implies a leak of a specific kind of virus; one that is not merely adapted to human cells but to transmission between whole, intact, humans. Only one theory of how SARS-CoV-2 arose fits this description. It is the Mojiang Miners Passage theory.
Where now for origin research?
Major institutions, primarily the EcoHealth Alliance, the NIH, and the WIV, hold large troves of information that could prove or disprove a lab leak. All assert their transparency and accountability, but, in practice, each has denied numerous requests for documents and other data. If one only listened to their words, one would think they wanted to find the origins of COVID-19, but their actions speak louder. Progress will therefore likely depend on independent initiatives.
When Jesse Bloom ingeniously retrieved previously deleted early SARS-CoV-2 sequences from the cloud and thereby recovered a novel early virus strain (Bloom, 2021), virologist Rasmus Nielsen said that this information was:
“the most important data that we have received regarding the origins of COVID-19 for more than a year”.
Nielsen was proven correct since Bloom’s discovery has been crucial to the improved rooting of Kumar and colleagues (Caraballo-Ortiz, et al., 2022).
The exceptional research value of these early sequences indicates also the appropriate scientific benchmark for judging those who erase or withold such evidence. History is full of ironies, but not many exceed the spectacle of prominent scientists, whose careers are publicly funded on the promise of identifying causes of infectious diseases, not wanting others to know the cause of the COVID-19 pandemic. Why, after all, did public institutions like NIH, NIAID, USAID, DOD, who have showered money on the EcoHealth Alliance, supposedly to prevent pandemics, not fund widespread searches for early SARS-CoV-2 infections at the very start of the pandemic?
Given this blockade, even at this late date, probably the simplest and most useful way to advance the scientific search for the origin of SARS-CoV-2 would be to locate and analyse more samples from early in the outbreak, from any country, regardless (Basavaraju et al., 2021; Montomoli et al; 2021; Canuti et al., 2022). Preliminary research suggests a plethora of suitable clinical and environmental samples exist in civilian and also military sample collections and databases, and that searches for early sequences are likely to be fruitful (Canuti et al. 2022, Paixao et al. 2022, Althoff et al. 2021; Chapleau et al. 2021; Lednicky et al. 2021; Basavaraju et al. 2021; Chen et al. 2020).
The other necessary benchmark is an ethical one. Dr Tedros of the WHO has called investigating the origin of the pandemic “a moral obligation“. We would go further. Although there is no law against obstructing origin research, it should nevertheless be considered a crime against all humanity to make it more likely that an event that resulted in millions of deaths and untold misery will recur because we never found its cause (Relman, 2020).
References
Andersen, K. G., Rambaut, A., Lipkin, W. I., Holmes, E. C., & Garry, R. F. (2020). The proximal origin of SARS-CoV-2. Nature medicine, 26(4), 450-452.Bloom, J. D. (2021). Recovery of deleted deep sequencing data sheds more light on the early Wuhan SARS-CoV-2 epidemic. Molecular biology and evolution, 38(12), 5211-5224.
Althoff, K. N., Schlueter, D. J., Anton-Culver, H., Cherry, J., Denny, J. C., Thomsen, I., … & Schully, S. D. (2022). Antibodies to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in All of Us Research Program participants, 2 January to 18 March 2020. Clinical Infectious Diseases, 74(4), 584-590.
Basavaraju, S. V., Patton, M. E., Grimm, K., Rasheed, M. A. U., Lester, S., Mills, L., … & Stramer, S. L. (2021). Serologic testing of US blood donations to identify severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)–reactive antibodies: December 2019–January 2020. Clinical Infectious Diseases, 72(12), e1004-e1009.
Caraballo-Ortiz, M. et al., (2022) TopHap: Rapid inference of key phylogenetic structures from common haplotypes in large genome collections with limited diversity. Bioinformatics, btac186
Chapleau, R. R., Christian, M., Connors, B., Premo, C., Chao, T. C., Rodriguez, J., … & Starr, C. (2021). Early Identification of SARS-CoV-2 Emergence in the DoD via Retrospective Analysis of 2019-2020 Upper Respiratory Illness Samples. medRxiv.
Courtier-Orgogozo, V., & de Ribera, F. A. (2022). SARS-CoV-2 infection at the Huanan seafood market. Zenodo.
Chen, C., Li, J., Di, L., Jing, Q., Du, P., Song, C., … & Wang, J. (2020). MINERVA: a facile strategy for SARS-CoV-2 whole-genome deep sequencing of clinical samples. Molecular cell, 80(6), 1123-1134.
Dearlove, B., Lewitus, E., Bai, H., Li, Y., Reeves, D. B., Joyce, M. G., … & Rolland, M. (2020). A SARS-CoV-2 vaccine candidate would likely match all currently circulating variants. Proceedings of the National Academy of Sciences, 117(38), 23652-23662.
Dudas, G., Carvalho, L. M., Rambaut, A., & Bedford, T. (2018). MERS-CoV spillover at the camel-human interface. elife, 7, e31257.
Frutos, R., Serra-Cobo, J., Chen, T., & Devaux, C. A. (2020). COVID-19: Time to exonerate the pangolin from the transmission of SARS-CoV-2 to humans. Infection, Genetics and Evolution, 84, 104493.
Gao, G., Liu, W., Liu, P., Lei, W., Jia, Z., He, X., … & Wu, G. (2022). Surveillance of SARS-CoV-2 in the environment and animal samples of the Huanan Seafood Market.
Gu, H., Chen, Q., Yang, G., He, L., Fan, H., Deng, Y. Q., … & Zhou, Y. (2020). Adaptation of SARS-CoV-2 in BALB/c mice for testing vaccine efficacy. Science, 369(6511), 1603-1607.
Kock, R., and Caceres-Escobar, H. (2022) Situation analysis on the roles and risks of wildlife in the emergence of human infectious diseases. IUCN
Kumar, S., Tao, Q., Weaver, S., Sanderford, M., Caraballo-Ortiz, M. A., Sharma, S., … & Miura, S. (2021). An evolutionary portrait of the progenitor SARS-CoV-2 and its dominant offshoots in COVID-19 pandemic. Molecular Biology and Evolution, 38(8), 3046-3059.
Latinne, A., Hu, B., Olival, K. J., Zhu, G., Zhang, L., Li, H., … & Daszak, P. (2020). Origin and cross-species transmission of bat coronaviruses in China. Nature Communications, 11(1), 1-15.
Li, H., Mendelsohn, E., Zong, C., Zhang, W., Hagan, E., Wang, N., … & Daszak, P. (2019). Human-animal interactions and bat coronavirus spillover potential among rural residents in Southern China. Biosafety and health, 1(02), 84-90.
Lednicky, J., Salemi, M., Subramaniam, K., Waltzek, T. B., Sabo-Attwood, T., Loeb, J. C., … & Morris Jr, J. G. (2021). Earliest detection to date of SARS-CoV-2 in Florida: Identification together with influenza virus on the main entry door of a university building, February 2020. Plos one, 16(1), e0245352.
Liu, Q., Zhao, S., Shi, C. M., Song, S., Zhu, S., Su, Y., … & Chen, H. (2020). Population genetics of SARS-CoV-2: disentangling effects of sampling bias and infection clusters. Genomics, proteomics & bioinformatics, 18(6), 640-647.
Manolio, T. A., Collins, F. S., Cox, N. J., Goldstein, D. B., Hindorff, L. A., Hunter, D. J., … & Visscher, P. M. (2009). Finding the missing heritability of complex diseases. Nature, 461(7265), 747-753.
Miura, S., Huuki, L. A., Buturla, T., Vu, T., Gomez, K., & Kumar, S. (2018). Computational enhancement of single-cell sequences for inferring tumor evolution. Bioinformatics, 34(17), i917-i926.
Montagutelli, X., Prot, M., Levillayer, L., Salazar, E. B., Jouvion, G., Conquet, L., … & Simon-Loriere, E. (2021). The B1. 351 and P. 1 variants extend SARS-CoV-2 host range to mice. BioRxiv.
Morel, B., Barbera, P., Czech, L., Bettisworth, B., Hübner, L., Lutteropp, S., … & Stamatakis, A. (2021). Phylogenetic analysis of SARS-CoV-2 data is difficult. Molecular biology and evolution, 38(5), 1777-1791.
Mostefai, F., Gamache, I., N’Guessan, A., Pelletier, J., Huang, J., Murall, C. L., … & Hussin, J. (2022). Population genomics approaches for genetic characterization of SARS-CoV-2 lineages. Frontiers in medicine, 207.
Paixao, J., Galangue, M., Gaston, C., Carralero, R., Lino, C., Júlio, G., … & Francisco, N. M. (2022). Early Evidence of Circulating SARS-CoV-2 in Unvaccinated and Vaccinated Measles Patients, September 2019–February 2020. Infection and Drug Resistance, 15, 533.
Pekar, J., Worobey, M., Moshiri, N., Scheffler, K., & Wertheim, J. O. (2021). Timing the SARS-CoV-2 index case in Hubei province. Science, 372(6540), 412-417.
Pekar, J., et al., (2022) SARS-CoV-2 emergence very likely resulted from at least two zoonotic events. Zenodo.
Pipes, L., Wang, H., Huelsenbeck, J. P., & Nielsen, R. (2021). Assessing uncertainty in the rooting of the SARS-CoV-2 phylogeny. Molecular biology and evolution, 38(4), 1537-1543.
Relman, D. A. (2020). Opinion: To stop the next pandemic, we need to unravel the origins of COVID-19. Proceedings of the National Academy of Sciences, 117(47), 29246-29248.
Schrago, C. G., & Barzilai, L. P. (2021). Challenges in estimating virus divergence times in short epidemic timescales with special reference to the evolution of SARS-CoV-2 pandemic. Genetics and molecular biology, 44.
Song, N., Cui, G. L., & Zeng, Q. L. (2021). Genomic epidemiology of SARS-CoV-2 from Mainland China. Frontiers in microbiology, 12, 1211.
van Dorp, L., Richard, D., Tan, C., Shaw, L. P., Acman, M., & Balloux, F. (2020). No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nature communications, 11(1), 1-8.
Tan, C., Lam, S. D., Richard, D., Owen, C., Berchtold, D., Orengo, C., … & Balloux, F. (2022). Transmission of SARS-CoV-2 from humans to animals and potential host adaptation.
Worobey et al. (2022) The Huanan market was the epicenter of SARS-CoV-2 emergence. Zenodo.
Xia, X. (2021). Dating the Common Ancestor from an NCBI Tree of 83688 High-Quality and Full-Length SARS-CoV-2 Genomes. Viruses, 13(9), 1790.
Zhan, S. H., Deverman, B. E., & Chan, Y. A. (2020). SARS-CoV-2 is well adapted for humans. What does this mean for re-emergence?. BioRxiv.
If this article was useful to you please consider sharing it with your networks.

Several of the references mentioned in the second-last paragraph are missing from the reference list: “(Canuti et al. 2022, Paixao et al. 2022, Althoff et al. 2021, Chapleau et al. 2021, Lednicky et al. 2021, … Chen et al. 2020)”; likewise Tan et al., 2022.
Fixed now, thank you.
Very good. What do you make of the mysterious “evali” and nursing home respiratory disease wave in in the summer of 2019 in the USA?
https://www.researchgate.net/publication/359231017_Is_SARS-CoV-2_the_causative_agent_of_EVALI_cases_prior_to_the_Covid-19_pandemic
https://therequestor.substack.com/p/is-wuhan-a-red-herring